systemPipeR: Workflow Design and Reporting Environment
35 minute read
Introduction
systemPipeR
provides flexible utilities for designing, building and running automated end-to-end
analysis workflows for a wide range of research applications, including
next-generation sequencing (NGS) experiments, such as RNA-Seq, ChIP-Seq,
VAR-Seq and Ribo-Seq (H Backman and Girke 2016). Important features include a uniform
workflow interface across different data analysis applications, automated
report generation, and support for running both R and command-line software,
such as NGS aligners or peak/variant callers, on local computers or compute
clusters (Figure 1). The latter supports interactive job submissions and batch
submissions to queuing systems of clusters.
systemPipeR has been designed to improve the reproducibility of large-scale data analysis
projects while substantially reducing the time it takes to analyze complex omics
data sets. It provides a uniform workflow interface and management system that allows
the users to run selected workflow steps, as well as customize and design entirely new workflows.
Additionally, the package take advantage of central community S4 classes of the Bioconductor
ecosystem, and enhances them with command-line software support.
The main motivation and advantages of using systemPipeR for complex data analysis tasks are:
- Design of complex workflows involving multiple R/Bioconductor packages
- Common workflow interface for different applications
- User-friendly access to widely used Bioconductor utilities
- Support of command-line software from within R
- Reduced complexity of using compute clusters from R
- Accelerated runtime of workflows via parallelization on computer systems with multiple CPU cores and/or multiple nodes
- Improved reproducibility by automating the generation of analysis reports
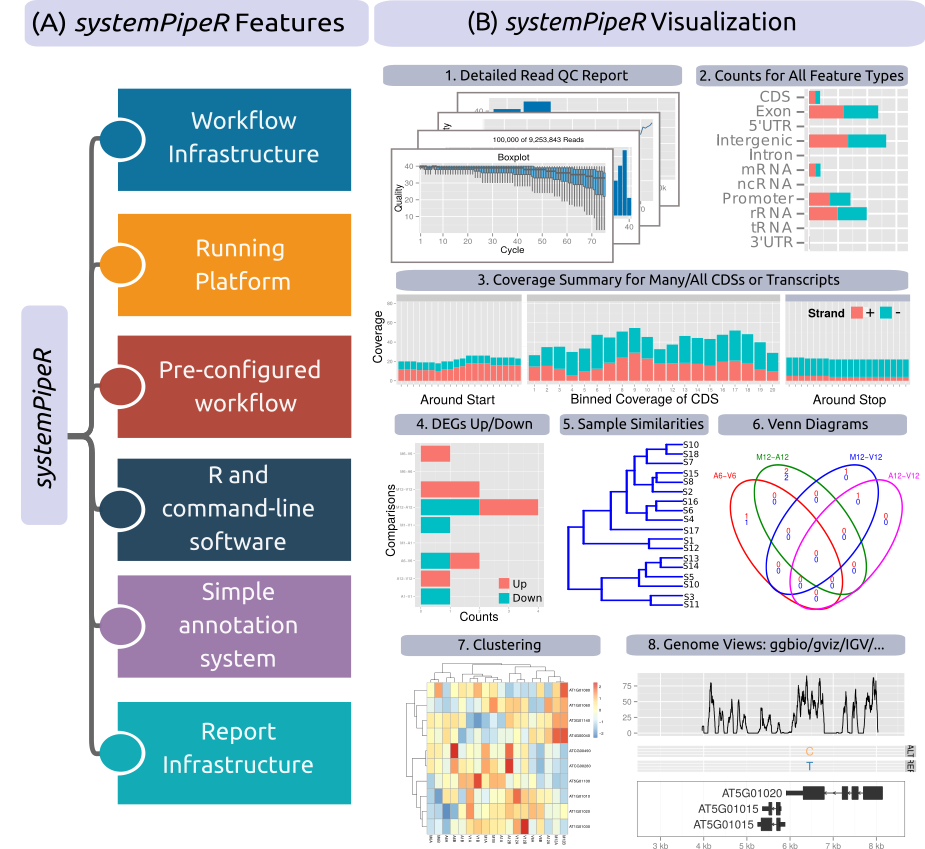
Figure 1: Relevant features in systemPipeR.
Workflow design concepts are illustrated under (A). Examples of systemPipeR's visualization functionalities are given under (B).
A central concept for designing workflows within the systemPipeR
environment is the use of workflow management containers. Workflow management
containers facilitate the design and execution of complex data
analysis steps. For its command-line interface systemPipeR adopts the
widely used Common Workflow Language (CWL)
(Amstutz et al. 2016). The interface to CWL is established by systemPipeR's
workflow control class called SYSargsList (see Figure 2). This
design offers many advantages such as: (i) options to run workflows either
entirely from within R, from various command-line wrappers (e.g., cwl-runner)
or from other languages (e.g., Bash or Python). Apart from providing
support for both command-line and R/Bioconductor software, the package provides
utilities for containerization, parallel evaluations on computer clusters and
automated generation of interactive analysis reports.
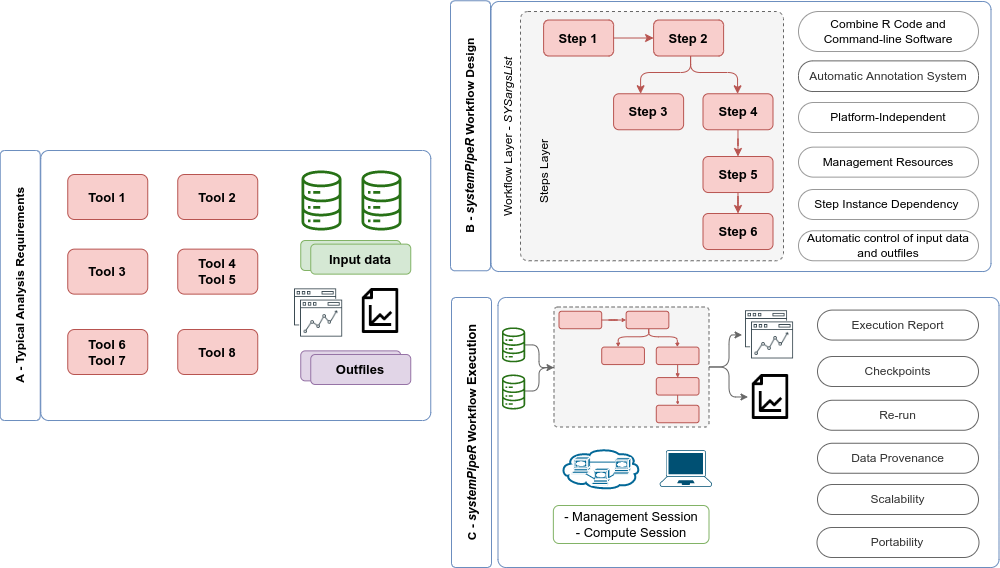
Figure 2: Overview of systemPipeR workflows management instances. (A) A
typical analysis workflow requires multiple software tools (red), as well the
description of the input (green) and output files, including analysis reports
(purple). (B) systemPipeR provides multiple utilities to design and
build a workflow, allowing multi-instance, integration of R code and command-line
software, a simple and efficient annotation system, that allows automatic control
of the input and output data, and multiple resources to manage the entire workflow.
(C) Options are provided to execute single or multiple workflow steps, while
enabling scalability, checkpoints, and generation of technical and scientific reports.
An important feature of systemPipeR's CWL interface is that it provides two
options to run command-line tools and workflows based on CWL. First, one can
run CWL in its native way via an R-based wrapper utility for cwl-runner or
cwl-tools (CWL-based approach). Second, one can run workflows using CWL’s
command-line and workflow instructions from within R (R-based approach). In the
latter case the same CWL workflow definition files (e.g. *.cwl and *.yml)
are used but rendered and executed entirely with R functions defined by
systemPipeR, and thus use CWL mainly as a command-line and workflow
definition format rather than execution software to run workflows. In this regard
systemPipeR also provides several convenience functions that are useful for
designing and debugging workflows, such as a command-line rendering function to
retrieve the exact command-line strings for each data set and processing step
prior to running a command-line.
Workflow Management with SYSargsList
The SYSargsList S4 class is a list-like container that stores the paths to
all input and output files along with the corresponding parameters used in each
analysis step (see Figure 3). SYSargsList instances are constructed from an
optional targets files, and two CWL parameter files including *.cwl and
*.yml (for details, see below). When running preconfigured NGS workflows, the
only input the user needs to provide is the initial targets file containing the
paths to the input files (e.g., FASTQ) and experiment design information, such
as sample labels and biological replicates. Subsequent targets instances are
created automatically, based on the connectivity establish between each
workflow step. SYSargsList containers store all information required for
one or multiple steps. This establishes central control for running, monitoring
and debugging complex workflows from start to finish.
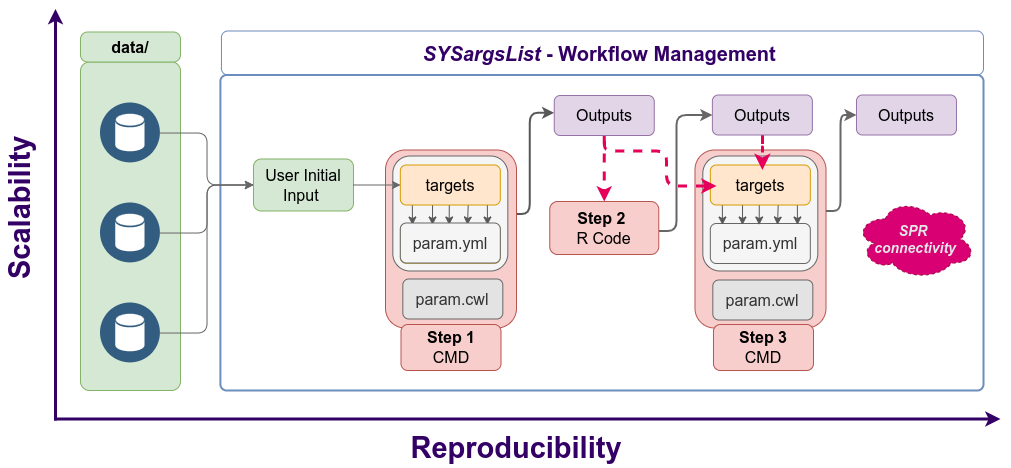
Figure 3: Workflow steps with input/output file operations are controlled by
the SYSargsList container. Each of its components (SYSargs2) are constructed
from an optional targets and two param files. Alternatively, LineWise instances
containing pure R code can be used.
Getting Started
Installation
The R software for running
systemPipeR
can be downloaded from CRAN. The
systemPipeR environment can be installed from the R console using the
BiocManager::install
command. The associated data package
systemPipeRdata
can be installed the same way. The latter is a helper package for generating
systemPipeR workflow environments with a single command containing all
parameter files and sample data required to quickly test and run workflows.
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("systemPipeR")
BiocManager::install("systemPipeRdata")
To use command-line software, the corresponding tool and dependencies need to be installed on a user’s system. See details.
Loading package and documentation
library("systemPipeR") # Loads the package
library(help = "systemPipeR") # Lists package info
vignette("systemPipeR") # Opens vignette
Load sample data and workflow templates
The mini sample FASTQ files used by this introduction as well as the
associated workflow reporting vignettes can be loaded via the
systemPipeRdata package as shown below. The chosen data set
SRP010938 contains 18
paired-end (PE) read sets from Arabidposis thaliana (Howard et al. 2013). To
minimize processing time during testing, each FASTQ file has been subsetted to
90,000-100,000 randomly sampled PE reads that map to the first 100,000
nucleotides of each chromosome of the A. thalina genome. The corresponding
reference genome sequence (FASTA) and its GFF annotation files (provided in the
same download) have been truncated accordingly. This way the entire test sample
data set requires less than 200MB disk storage space. A PE read set has been
chosen for this test data set for flexibility, because it can be used for
testing both types of analysis routines requiring either SE (single-end) reads
or PE reads.
The following generates a fully populated systemPipeR workflow environment
(here for RNA-Seq) in the current working directory of an R session. The systemPipeRdata
package provides preconfigured workflow templates for RNA-Seq, ChIP-Seq, VAR-Seq, and Ribo-Seq.
Additional, templates are available on the project web site here.
systemPipeRdata::genWorkenvir(workflow = "rnaseq")
setwd("rnaseq")
Project structure
The working environment of the sample data loaded in the previous step contains the following pre-configured directory structure (Figure 4). Directory names are indicated in green. Users can change this structure as needed, but need to adjust the code in their workflows accordingly.
- workflow/ (e.g. rnaseq/)
- This is the root directory of the R session running the workflow.
- Run script ( *.Rmd) and sample annotation (targets.txt) files are located here.
- Note, this directory can have any name (e.g. rnaseq, varseq). Changing its name does not require any modifications in the run script(s).
- Important subdirectories:
- param/
- param/cwl/: This subdirectory stores all the CWL parameter files. To organize workflows, each can have its own subdirectory, where all
CWL paramandinput.ymlfiles need to be in the same subdirectory.
- param/cwl/: This subdirectory stores all the CWL parameter files. To organize workflows, each can have its own subdirectory, where all
- data/
- FASTQ files
- FASTA file of reference (e.g. reference genome)
- Annotation files
- etc.
- results/
- Analysis results are usually written to this directory, including: alignment, variant and peak files (BAM, VCF, BED); tabular result files; and image/plot files.
- Note, the user has the option to organize results files for a given sample and analysis step in a separate subdirectory.
- param/
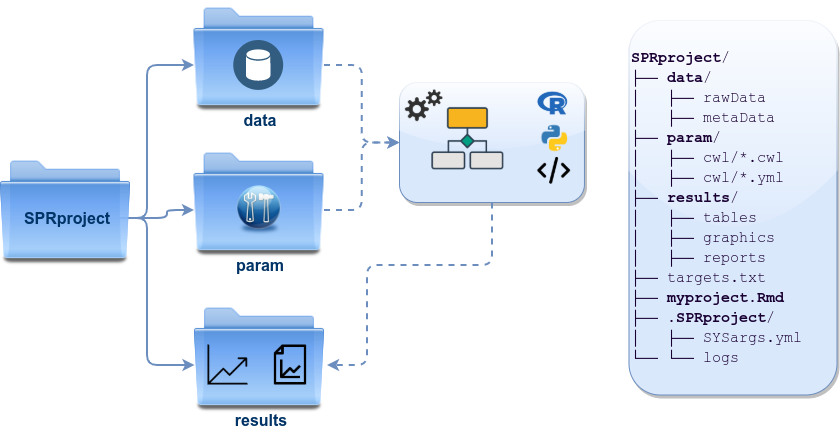
Figure 4: systemPipeR’s preconfigured directory structure.
The following parameter files are included in each workflow template:
targets.txt: initial one provided by user; downstreamtargets_*.txtfiles are generated automatically*.param/cwl: defines parameter for input/output file operations, e.g.:hisat2/hisat2-mapping-se.cwlhisat2/hisat2-mapping-se.yml
- Configuration files for computer cluster environments (skip on single machines):
.batchtools.conf.R: defines the type of scheduler forbatchtoolspointing to template file of cluster, and located in user’s home directory*.tmpl: specifies parameters of scheduler used by a system, e.g. Torque, SGE, Slurm, etc.
Structure of targets file
The targets file defines all input files (e.g. FASTQ, BAM, BCF) and
sample comparisons of an analysis workflow. The following shows the format of a
sample targets file included in the package. It also can be viewed and
downloaded from systemPipeR’s GitHub repository
here.
In a target file with a single type of input files, here FASTQ files of
single-end (SE) reads, the first three columns are mandatory including their
column names, while it is four mandatory columns for FASTQ files of PE reads.
All subsequent columns are optional and any number of additional columns can be
added as needed. The columns in targets files are expected to be tab separated (TSV format).
The SampleName column contains usually short labels for
referencing samples (here FASTQ files) across many workflow steps (e.g.
plots and column titles). Importantly, the labels used in the SampleName
column need to be unique, while technical or biological replicates are
indicated by duplicated values under the Factor column. For readability
and transparency, it is useful to use here a short, consistent and informative
syntax for naming samples and replicates. To avoid problems with other
packages or external software, it is recommended to use the basic naming rules
for R objects and their components as outlined here.
This is important since the values used under the SampleName and Factor
columns are intended to be used as labels for naming columns or plotting features
in downstream analysis steps.
Users should note here, the usage of targets files is optional when using systemPipeR’s new CWL interface. They can be replaced by a standard YAML input file used by CWL. Since for organizing experimental variables targets files are extremely useful and user-friendly. Thus, we encourage users to keep using them.
Structure of targets file for single-end (SE) samples
library(systemPipeR)
targetspath <- system.file("extdata", "targets.txt", package = "systemPipeR")
showDF(read.delim(targetspath, comment.char = "#"))
To work with custom data, users need to generate a targets file containing
the paths to their own FASTQ files and then provide under targetspath the
path to the corresponding targets file.
Structure of targets file for paired-end (PE) samples
For paired-end (PE) samples, the structure of the targets file is similar, where
users need to provide two FASTQ path columns: FileName1 and FileName2
with the paths to the PE FASTQ files.
targetspath <- system.file("extdata", "targetsPE.txt", package = "systemPipeR")
showDF(read.delim(targetspath, comment.char = "#"))
Sample comparisons
Sample comparisons are defined in the header lines of the targets file
starting with ‘# <CMP>’.
readLines(targetspath)[1:4]
## [1] "# Project ID: Arabidopsis - Pseudomonas alternative splicing study (SRA: SRP010938; PMID: 24098335)"
## [2] "# The following line(s) allow to specify the contrasts needed for comparative analyses, such as DEG identification. All possible comparisons can be specified with 'CMPset: ALL'."
## [3] "# <CMP> CMPset1: M1-A1, M1-V1, A1-V1, M6-A6, M6-V6, A6-V6, M12-A12, M12-V12, A12-V12"
## [4] "# <CMP> CMPset2: ALL"
The function readComp imports the comparison information and stores it in a
list. Alternatively, readComp can obtain the comparison information from
the corresponding SYSargsList object (see below). Note, these header lines are
optional. They are mainly useful for controlling comparative analyses according
to certain biological expectations, such as identifying differentially expressed
genes in RNA-Seq experiments based on simple pair-wise comparisons.
readComp(file = targetspath, format = "vector", delim = "-")
## $CMPset1
## [1] "M1-A1" "M1-V1" "A1-V1" "M6-A6" "M6-V6" "A6-V6" "M12-A12"
## [8] "M12-V12" "A12-V12"
##
## $CMPset2
## [1] "M1-A1" "M1-V1" "M1-M6" "M1-A6" "M1-V6" "M1-M12" "M1-A12"
## [8] "M1-V12" "A1-V1" "A1-M6" "A1-A6" "A1-V6" "A1-M12" "A1-A12"
## [15] "A1-V12" "V1-M6" "V1-A6" "V1-V6" "V1-M12" "V1-A12" "V1-V12"
## [22] "M6-A6" "M6-V6" "M6-M12" "M6-A12" "M6-V12" "A6-V6" "A6-M12"
## [29] "A6-A12" "A6-V12" "V6-M12" "V6-A12" "V6-V12" "M12-A12" "M12-V12"
## [36] "A12-V12"
Project initialization
To create a workflow within systemPipeR, we can start by defining an empty
container and checking the directory structure:
sal <- SPRproject()
## Creating directory '/home/tgirke/tmp/GEN242/content/en/tutorials/systempiper/rnaseq/.SPRproject'
## Creating file '/home/tgirke/tmp/GEN242/content/en/tutorials/systempiper/rnaseq/.SPRproject/SYSargsList.yml'
Internally, SPRproject function will create a hidden directory called .SPRproject,
by default. This directory will store all log files generated during a workflow
run.
Initially, the object sal is a empty container containing only the basic project
information. The project information can be accessed with the projectInfo method.
sal
## Instance of 'SYSargsList':
## No workflow steps added
projectInfo(sal)
## $project
## [1] "/home/tgirke/tmp/GEN242/content/en/tutorials/systempiper/rnaseq"
##
## $data
## [1] "data"
##
## $param
## [1] "param"
##
## $results
## [1] "results"
##
## $logsDir
## [1] ".SPRproject"
##
## $sysargslist
## [1] ".SPRproject/SYSargsList.yml"
Also, the length function will return how many steps a workflow contains. Since
no steps have been added yet it returns zero.
length(sal)
## [1] 0
Building workflows
systemPipeR workflows can be populated with a single command from an R
Markdown file or stepwise in interactive mode. This section introduces first
how to build a workflow stepwise in interactive mode, and then how to
build a workflow with a single command from an R Markdown file.
Stepwise workflow construction
New workflows are constructed, or existing ones modified, by connecting each
step via the appendStep method. Each step in a SYSargsList instance contains the
instructions needed for processing a set of input files with a specific
command-line and the paths to the exptected outfiles. For constructing R code-based
workflow steps the constructor function Linewise is used.
The following demonstrates how to create a command-line workflow step here using as example the short read aligner software HISAT2 (Kim, Langmead, and Salzberg 2015).
The constructor function renders the proper command-line strings for each sample
and software tool, appending a new step in the SYSargsList object defined in the
previous step. For this, the SYSargsList constructor function uses in this example
data from three input files:
- CWL command-line specification file (
wf_fileargument) - Input variables (
input_fileargument) - Targets file (
targetsargument)
In CWL files with the extension .cwl define the parameters of a chosen
command-line step or an entire workflow, while files with the extension .yml define
the input variables of command-line steps. Note, input variables provided
by a targets or targetsPE file can be passed on via the inputvars argument to
the .yml file and from there to the SYSargsList object.
appendStep(sal) <- SYSargsList(step_name = "hisat2_mapping", dir = TRUE, targets = "targetsPE.txt",
wf_file = "workflow-hisat2/workflow_hisat2-pe.cwl", input_file = "workflow-hisat2/workflow_hisat2-pe.yml",
dir_path = "param/cwl", inputvars = c(FileName1 = "_FASTQ_PATH1_", FileName2 = "_FASTQ_PATH2_",
SampleName = "_SampleName_"))
An overview of the workflow can be printed as follows.
sal
## Instance of 'SYSargsList':
## WF Steps:
## 1. hisat2_mapping --> Status: Pending
## Total Files: 72 | Existing: 0 | Missing: 72
## 1.1. hisat2
## cmdlist: 18 | Pending: 18
## 1.2. samtools-view
## cmdlist: 18 | Pending: 18
## 1.3. samtools-sort
## cmdlist: 18 | Pending: 18
## 1.4. samtools-index
## cmdlist: 18 | Pending: 18
##
In the above the workflow status for the new step is Pending. This means the workflow object has been constructed but not executed yet.
Several accessor methods are available to explore the SYSargsList object.
Of particular interest is the cmdlist() method. It constructs the system
commands for running command-line software as specified by a given .cwl
file combined with the paths to the input samples (e.g. FASTQ files), here provided
by a targets file. The example below shows the cmdlist() output for
running HISAT2 on the first PE read sample. Evaluating the output of
cmdlist() can be very helpful for designing and debugging .cwl files
of new command-line software or changing the parameter settings of existing
ones. The rendered command-line instance for each input sample can be returned as follows.
cmdlist(sal, step = "hisat2_mapping", targets = 1)
## $hisat2_mapping
## $hisat2_mapping$M1A
## $hisat2_mapping$M1A$hisat2
## [1] "hisat2 -S ./results/M1A.sam -x ./data/tair10.fasta -k 1 --min-intronlen 30 --max-intronlen 3000 -1 ./data/SRR446027_1.fastq.gz -2 ./data/SRR446027_2.fastq.gz --threads 4"
##
## $hisat2_mapping$M1A$`samtools-view`
## [1] "samtools view -bS -o ./results/M1A.bam ./results/M1A.sam "
##
## $hisat2_mapping$M1A$`samtools-sort`
## [1] "samtools sort -o ./results/M1A.sorted.bam ./results/M1A.bam -@ 4"
##
## $hisat2_mapping$M1A$`samtools-index`
## [1] "samtools index -b results/M1A.sorted.bam results/M1A.sorted.bam.bai ./results/M1A.sorted.bam "
The outfiles components of SYSargsList define the expected output files for
each step in the workflow; some of which are the input for the next workflow step
(see Figure 3).
outfiles(sal)
## $hisat2_mapping
## DataFrame with 18 rows and 4 columns
## hisat2_sam samtools_bam samtools_sort_bam
## <character> <character> <character>
## M1A ./results/M1A.sam ./results/M1A.bam ./results/M1A.sorted..
## M1B ./results/M1B.sam ./results/M1B.bam ./results/M1B.sorted..
## A1A ./results/A1A.sam ./results/A1A.bam ./results/A1A.sorted..
## A1B ./results/A1B.sam ./results/A1B.bam ./results/A1B.sorted..
## V1A ./results/V1A.sam ./results/V1A.bam ./results/V1A.sorted..
## ... ... ... ...
## M12B ./results/M12B.sam ./results/M12B.bam ./results/M12B.sorte..
## A12A ./results/A12A.sam ./results/A12A.bam ./results/A12A.sorte..
## A12B ./results/A12B.sam ./results/A12B.bam ./results/A12B.sorte..
## V12A ./results/V12A.sam ./results/V12A.bam ./results/V12A.sorte..
## V12B ./results/V12B.sam ./results/V12B.bam ./results/V12B.sorte..
## samtools_index
## <character>
## M1A ./results/M1A.sorted..
## M1B ./results/M1B.sorted..
## A1A ./results/A1A.sorted..
## A1B ./results/A1B.sorted..
## V1A ./results/V1A.sorted..
## ... ...
## M12B ./results/M12B.sorte..
## A12A ./results/A12A.sorte..
## A12B ./results/A12B.sorte..
## V12A ./results/V12A.sorte..
## V12B ./results/V12B.sorte..
In an ‘R-centric’ rather than a ‘CWL-centric’ workflow design the connectivity
among workflow steps is established by creating the downstream targets instances
automatically (see Figure 3). Each step uses the outfiles from the previous step as
input, thereby establishing connectivity among each step in the workflow. By chaining
several SYSargsList steps together one can construct complex workflows involving
many sample-level input/output file operations with any combination of command-line or R-based
software. Also, systemPipeR provides features to automatically build these
connections. This provides additional security to make sure that all samples will be
processed in a reproducible manner.
Alternatively, a CWL-centric workflow design can be used that defines all or most workflow steps with CWL workflow and parameter files. Due to time and space restrictions, the CWL-centric approach is not covered by this tutorial.
The following R code step generates a data.frame containing important read alignment statistics
such as the total number of reads in the FASTQ files, the number of total alignments,
as well as the number of primary alignments in the corresponding BAM files.
The constructor function LineWise requires the step_name and the R code
assigned to the code argument. The R code should be enclosed by braces ({}).
appendStep(sal) <- LineWise(code = {
fqpaths <- getColumn(sal, step = "hisat2_mapping", "targetsWF", column = "FileName1")
bampaths <- getColumn(sal, step = "hisat2_mapping", "outfiles", column = "samtools_sort_bam")
read_statsDF <- alignStats(args = bampaths, fqpaths = fqpaths, pairEnd = TRUE)
write.table(read_statsDF, "results/alignStats.xls", row.names = FALSE, quote = FALSE,
sep = "\t")
}, step_name = "align_stats", dependency = "hisat2_mapping")
To inspect the R code of this step, the codeLine method can be used.
codeLine(sal, "align_stats")
## align_stats
## fqpaths <- getColumn(sal, step = "hisat2_mapping", "targetsWF", column = "FileName1")
## bampaths <- getColumn(sal, step = "hisat2_mapping", "outfiles", column = "samtools_sort_bam")
## read_statsDF <- alignStats(args = bampaths, fqpaths = fqpaths, pairEnd = TRUE)
## write.table(read_statsDF, "results/alignStats.xls", row.names = FALSE, quote = FALSE, sep = "\t")
The getColumn method allows to extract certain information of a workflow instance, here
output files that can be accessed from within R as demonstrated below.
getColumn(sal, step = "hisat2_mapping", "outfiles", column = "samtools_sort_bam")
## M1A M1B
## "./results/M1A.sorted.bam" "./results/M1B.sorted.bam"
## A1A A1B
## "./results/A1A.sorted.bam" "./results/A1B.sorted.bam"
## V1A V1B
## "./results/V1A.sorted.bam" "./results/V1B.sorted.bam"
## M6A M6B
## "./results/M6A.sorted.bam" "./results/M6B.sorted.bam"
## A6A A6B
## "./results/A6A.sorted.bam" "./results/A6B.sorted.bam"
## V6A V6B
## "./results/V6A.sorted.bam" "./results/V6B.sorted.bam"
## M12A M12B
## "./results/M12A.sorted.bam" "./results/M12B.sorted.bam"
## A12A A12B
## "./results/A12A.sorted.bam" "./results/A12B.sorted.bam"
## V12A V12B
## "./results/V12A.sorted.bam" "./results/V12B.sorted.bam"
Loading workflows from an R Markdown
The importWF function allows to load an entire workflow from an R Markdown (Rmd) file
into an SYSargsList object that has been intialized with SPRproject() as introduced
above. Next, one can run the workflow from start to finish with a single function call
using runWF. Below the latter has been commented out to first introduce additional
details prior to executing the workflow with runWF, which will be discussed in the
following section.
sal <- SPRproject()
## Creating directory '/home/tgirke/tmp/GEN242/content/en/tutorials/systempiper/rnaseq/.SPRproject'
## Creating file '/home/tgirke/tmp/GEN242/content/en/tutorials/systempiper/rnaseq/.SPRproject/SYSargsList.yml'
sal <- importWF(sal, file_path = "systemPipeRNAseq.Rmd")
## Reading Rmd file
##
## ---- Actions ----
## Ignore none-R chunks at line: 25
## Checking chunk eval values
## Checking chunk SPR option
## Ignore non-SPR chunks: 34, 43, 61, 96, 121, 188, 205, 298, 658, 686, 704, 712, 723
## Parse chunk code
## Now importing step 'load_SPR'
## Now importing step 'preprocessing'
## Now importing step 'trimming'
## Now importing step 'fastq_report'
## Now importing step 'hisat2_index'
## Now importing step 'hisat2_mapping'
## Now importing step 'align_stats'
## Now importing step 'bam_IGV'
## Now importing step 'create_db'
## Now importing step 'read_counting'
## Now importing step 'sample_tree'
## Now importing step 'run_edger'
## Now importing step 'custom_annot'
## Now importing step 'filter_degs'
## Now importing step 'venn_diagram'
## Now importing step 'get_go_annot'
## Now importing step 'go_enrich'
## Now importing step 'go_plot'
## Now importing step 'heatmap'
## Now importing step 'sessionInfo'
# sal <- runWF(sal)
Several accessor functions are available to inspect the workflow.
stepsWF(sal)
dependency(sal)
codeLine(sal)
targetsWF(sal)
Third-party software tools
Examples of preconfigured CWL templates for third-party software tools are provided in the following table. In addition, the tutorial entitled Automate Creation of CWL Instructions explains how to create these CWL templates for essentially any command-line tool simply by providing the command-line string for a new software as input.
|
Tool Name |
Description |
Step |
|---|---|---|
|
BWA is a software package for mapping low-divergent sequences against a large reference genome, such as the human genome. |
Alignment |
|
|
Bowtie 2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. |
Alignment |
|
|
FASTX-Toolkit is a collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing. |
Read Preprocessing |
|
|
Transrate is software for de-novo transcriptome assembly quality analysis. |
Quality |
|
|
GSNAP is a genomic short-read nucleotide alignment program. |
Alignment |
|
|
Samtools is a suite of programs for interacting with high-throughput sequencing data. |
Post-processing |
|
|
Trimmomatic is a flexible read trimming tool for Illumina NGS data. |
Read Preprocessing |
|
|
Rsubread is a Bioconductor software package that provides high-performance alignment and read counting functions for RNA-seq reads. |
Alignment |
|
|
Picard is a set of command line tools for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. |
Manipulating HTS data |
|
|
BUSCO assesses genome assembly and annotation completeness with Benchmarking Universal Single-Copy Orthologs. |
Quality |
|
|
HISAT2 is a fast and sensitive alignment program for mapping NGS reads (both DNA and RNA) to reference genomes. |
Alignment |
|
|
TopHat is a fast splice junction mapper for RNA-Seq reads. |
Alignment |
|
|
Variant Discovery in High-Throughput Sequencing Data. |
Variant Discovery |
|
|
STAR is an ultrafast universal RNA-seq aligner. |
Alignment |
|
|
Trim Galore is a wrapper around Cutadapt and FastQC to consistently apply adapter and quality trimming to FastQ files. |
Read Preprocessing |
|
|
TransDecoder identifies candidate coding regions within transcript sequences. |
Find Coding Regions |
|
|
Trinity assembles transcript sequences from Illumina RNA-Seq data. |
denovo Transcriptome Assembly |
|
|
Trinotate is a comprehensive annotation suite designed for automatic functional annotation of transcriptomes. |
Transcriptome Functional Annotation |
|
|
MACS2 identifies transcription factor binding sites in ChIP-seq data. |
Peak calling |
|
|
kallisto is a program for quantifying abundances of transcripts from RNA-Seq data. |
Read counting |
|
|
BCFtools is a program for variant calling and manipulating files in the Variant Call Format (VCF) and its binary counterpart BCF. |
Variant Discovery |
|
|
Bismark is a program to map bisulfite treated sequencing reads to a genome of interest and perform methylation calls in a single step. |
Bisulfite mapping |
|
|
FastQC is a quality control tool for high throughput sequence data. |
Quality |
|
|
BLAST finds regions of similarity between biological sequences. |
Blast |
Importantly, command-line software needs to be installed on a user’s system
and available in a user’s PATH. To check whether this is the case, one can use the
tryCL function.
tryCL(command = "grep")
How to run workflows?
Setup and requirements
To execute the code in this tutorial, one needs the following software installed.
- R (version >=4.1.2)
- systemPipeR package (version >=2.0.8)
- Hisat2 (version >= 2.1.0)
To build custom workflows with any additional command-line software, one needs
the respective software installed and available in the user PATH. To test this,
one can use the tryCL function.
tryCL(command = "hisat2") ## 'All set up, proceed!'
Run from R
For running a workflow, the runWF function can be used. It executes all workflow steps
stored in a SYSargsList container (below named sal). This assumes the sal object
has been initialized and populated as outlined above using SPRproject() and
importWF(sal, file_path="..."), respectively.
sal <- runWF(sal)
This essential function allows the user to choose one or multiple steps to be
executed using its steps argument. However, it is necessary to maintain valid
dependencies (also see workflow graph).
If a selected step depends on a previous step(s), the output of which may not
be available yet, then the execution will fail.
sal <- runWF(sal, steps = c(1, 3))
The runWF function allows to force the execution of chosen workflow steps, even if
the status of a given step is 'Success' and the expected outfiles exist already.
This is useful for updating certain steps when needed. Another option is to
force runWF to ignore all warnings and errors. This can be achieved by assigning
FALSE to the arguments warning.stop and error.stop, respectively.
sal <- runWF(sal, force = TRUE, warning.stop = FALSE, error.stop = FALSE)
While SYSargsList (sal) objects are autosaved when working with workflows, it can
be sometimes safer to explicity save the object before closing R.
sal <- write_SYSargsList(sal)
Resume and reset workflows
To restart workflows, set resume=TRUE.
sal <- SPRproject(resume = TRUE)
To delete all steps in a workflow including the content saved under the .SPRproject directory,
use overwrite=TRUE. This option should be used with caution, since it effectively deletes most
previous workflow data.
sal <- SPRproject(overwrite = TRUE)
For additional options and details, users want to consult the help
documentation for: ?SPRproject, ?runWF and ?'SYSargsList-class'.
Parallelization on clusters
The computation of time-consuming steps can be greatly accelerated by processing many files in parallel using several compute nodes of a cluster, where a scheduling system is used for load balancing.
The following resources list provides to a SYSargsList (sal) object the
number of independent parallel cluster processes defined under the Njobs
element in the list. The given example will run 18 processes in parallel using
for each 4 CPU cores, thus utilizing a total of 72 CPU cores. Note, runWF can
be used with most queueing systems as it is based on utilities defined by the
batchtools package, which supports the use of template files (*.tmpl) for
defining the run parameters of different schedulers. To run the following code,
one needs to have both a conffile (see .batchtools.conf.R samples
here) and a template file (see
*.tmpl samples
here) for the
queueing system available on a system. The following example uses the sample
conffile and template files for the Slurm scheduler provided by this
package.
The resources list can be appended when a workflow step is generated. Alternatively,
one can append these resource specifications to any step of a pre-generated SYSarsList
with the addResources function.
resources <- list(conffile=".batchtools.conf.R",
template="batchtools.slurm.tmpl",
Njobs=18,
walltime=120, # in min
ntasks=1,
ncpus=4,
memory=1024, # in Mb
partition = "short"
)
sal <- addResources(sal, c("hisat2_mapping"), resources = resources)
sal <- runWF(sal)
The above example will submit via runWF(sal) the hisat2_mapping step
to a partition called short on a computer cluster. Users need to adjust this and
other parameters defined in the resources list to their environment.
Run from Command-Line (without cluster)
Create an R script containing the following (or similar) minimum content.
#!/usr/bin/env Rscript
library(systemPipeR)
sal <- SPRproject()
sal <- importWF(sal, file_path = "systemPipeRNAseq.Rmd") # adjust name of Rmd file if different
sal <- runWF(sal) # runs entire workflow
sal <- renderReport(sal) # after workflow has completed render Rmd to HTML report
Assuming the script is named wf_run_script.R it can be executed from the command-line (not
R console!) as follows. In addition, one can make the script executable to run it like any other script.
Rscript wf_run_script.R
This will run systemPipeR workflows on a single machine. In this case a limited amount of
parallelization is possible if the corresponding code chunks in the workflow take advantage of
multi-core parallelization instructions provided by BiocParallel, batchtools or
related packages. However, this type of parallelization is usually limited to the
number of cores available on a single CPU. A much more scalable approach is the use
of computer clusters as described above and in the next section.
Submit workflow from command-line to cluster
In addition to running workflows within interactive R sessions or submitting
them from the command-line on a single system (see above), one can execute
systemPipeR workflows from the command-line to an HPC cluster by including
the relevant workflow run instructions in an R script and then submitting it
via a submission script of a workload manager system to a computer cluster. The
following gives an example for the Slurm workload manager. To understand the
details, it is important to inspect the content of the two files (here .R and
.sh). Additional details about resource specification under Slurm are given
below.
- R script: wf_run_script.R
- Slurm submission script: wf_run_script.sh
As a test, users can generate in their user account of a cluster a workflow
environment populated with the toy data as outlined
here.
After this, it is best to create within the workflow directory a subdirectory,
e.g. called cl_sbatch_run, and then save the above two files to this
subdirectory. Next, the parameters in both files need to be adjusted to match
the type of workflow and the required computing resources. This includes the
name of the Rmd file and scheduler resource settings such as: partition,
Njobs, walltime, memory, etc. After all relevant settings have been set
correctly, one can execute the workflow with sbatch from within the
cl_sbatch_run directory as follows (note this is a command-line call outside
of R):
sbatch wf_run_script.sh
After the submission to the cluster, one usually should check its status and
progress with squeue -u <username> (under Slurm) as well as by monitoring
the content of the slurm-<jobid>.out file generated by the scheduler in the
same directory. This file contains most of the STDOUT and STDERROR
generated by a cluster job. Once everything is working on the toy data, users
can run the workflow on real data the same way.
Visualize workflow
systemPipeR workflows instances can be visualized with the plotWF function.
The resulting plot includes the following information.
- Workflow topology graph (dependency graphs between different steps)
- Workflow step status, e.g.
Success,Error,Pending,Warnings - Sample status and statistics
- Workflow timing: run duration time
If no argument is provided, the basic plot will automatically detect width, height, layout, plot method, branches, etc.
plotWF(sal)
Technical reports
systemPipeR compiles all workflow execution logs in one central location,
making it easy to check any standard output (stdout) or standard error
(stderr) for any command-line tool used in a workflow.
Also, the information is appended to the workflow plot making it easy to click on
respective steps.
sal <- renderLogs(sal)
Scientific reports
systemPipeR auto-generates scientific analysis reports in HTML format. These reports
compile the results of all workflow steps including text, code, plots and tables.
sal <- renderReport(sal)
Alternatively, scientific reports can be rendered with the render function from rmarkdown.
rmarkdown::render("systemPipeRNAseq.Rmd", clean = TRUE, output_format = "BiocStyle::html_document")
How to modify workflows?
Modifying steps
If needed one can modify existing workflow steps in a pre-populated
SYSargsList object, and potentially already executed workflows, with the
replaceStep(sal) <- replacement function. The following gives an example
where step number 3 in a SYSargsList (sal) object will be updated with
modified or new code. Note, this is a generalized example where the user needs
to insert the code lines between { and }, and also adjust the values assigned
to the arguments: step_name and dependency.
replaceStep(sal, step = 3) <- LineWise(code = {
...
}, step_name = "my_step_name", dependency = "my_dependency")
Since step_names need to be unique, one should avoid using the same
step_name as before. If the previous name is used, a default name will be
assigned. Rerunning the assignment will then allow to assign the previous name. This
behavior is enforced for version tracking. Subsequently, one can view and check
the code changes with codeLine(), and then rerun the corresponding step (here
3) as follows:
codeLine(stepsWF(sal)$my_step_name)
runWF(sal, steps = 3)
As mentioned above, any step in a workflow can only be run in isolation if its
expected input exists (see dependency).
Adding steps
New steps can be added to the Rmd file of a workflow by inserting new R
Markdown code blocks (chunks) starting and ending with the usual appendStep<-
syntax and then creating a new SYSargsList instance with importWF that
contain the new step(s). To add steps to a pre-populated SYSargsList object,
one can use the after argument of the appendStep<- function. The following
example will add a new step after position 3 to the corresponding sal object.
This can be useful if a workflow with a long runtime has already been completed
and one only wants to make some refinements without re-running the entire
workflow.
appendStep(sal, after = 3) <- ...
Workflow templates
Workflow templates are provided via the affiliated Bioconductor data package
named systemPipeRdata, as well as by a dedicated GitHub repository. Instances
of these workflows can be created with a single command. The following gives
several examples.
RNA-Seq WF template
[ Tutorial 1 ] [ Tutorial 2 ]
Load the RNA-Seq sample workflow into your current working directory.
library(systemPipeRdata)
genWorkenvir(workflow = "rnaseq")
setwd("rnaseq")
Create the workflow
This template includes the most common analysis steps of RNAseq workflows. One can add, remove, modify
workflow steps by modifying the sal object.
sal <- SPRproject()
sal <- importWF(sal, file_path = "systemPipeRNAseq.Rmd", verbose = FALSE)
Workflow steps:
- Read preprocessing
- Quality filtering (trimming)
- FASTQ quality report
- Alignments:
HISAT2(or any other RNA-Seq aligner) - Alignment stats
- Read counting
- Sample-wise correlation analysis
- Analysis of differentially expressed genes (DEGs)
- GO term enrichment analysis
- Gene-wise clustering
Run workflow
sal <- runWF(sal)
Workflow visualization
plotWF(sal)
Report generation
sal <- renderReport(sal)
sal <- renderLogs(sal)
ChIP-Seq WF template
[ Tutorial 1 ] [ Tutorial 2 ]
Load the ChIP-Seq sample workflow into your current working directory.
library(systemPipeRdata)
genWorkenvir(workflow = "chipseq")
setwd("chipseq")
Workflow steps:
- Read preprocessing
- Quality filtering (trimming)
- FASTQ quality report
- Alignments:
Bowtie2orrsubread - Alignment stats
- Peak calling:
MACS2 - Peak annotation with genomic context
- Differential binding analysis
- GO term enrichment analysis
- Motif analysis
Create the workflow
This template provides some common steps for a ChIPseq workflow. One can add, remove, modify
workflow steps by operating on the sal object.
sal <- SPRproject()
sal <- importWF(sal, file_path = "systemPipeChIPseq.Rmd", verbose = FALSE)
Run workflow
sal <- runWF(sal)
Workflow visualization
plotWF(sal)
Report generation
sal <- renderReport(sal)
sal <- renderLogs(sal)
VAR-Seq WF template
[ Tutorial ]
Load the VAR-Seq sample workflow into your current working directory.
library(systemPipeRdata)
genWorkenvir(workflow = "varseq")
setwd("varseq")
Workflow steps:
- Read preprocessing
- Quality filtering (trimming)
- FASTQ quality report
- Alignments:
gsnap,bwa - Variant calling:
VariantTools,GATK,BCFtools - Variant filtering:
VariantToolsandVariantAnnotation - Variant annotation:
VariantAnnotation - Combine results from many samples
- Summary statistics of samples
Create the workflow
This template provides some common steps for a VARseq workflow. One can add, remove, modify
workflow steps by operating on the sal object.
sal <- SPRproject()
sal <- importWF(sal, file_path = "systemPipeVARseq.Rmd", verbose = FALSE)
Run workflow
sal <- runWF(sal)
Workflow visualization
plotWF(sal)
Report generation
sal <- renderReport(sal)
sal <- renderLogs(sal)
Ribo-Seq WF template
[ Tutorial ]
Load the Ribo-Seq sample workflow into your current working directory.
library(systemPipeRdata)
genWorkenvir(workflow = "riboseq")
setwd("riboseq")
Workflow steps:
- Read preprocessing
- Adaptor trimming and quality filtering
- FASTQ quality report
- Alignments:
HISAT2(or any other RNA-Seq aligner) - Alignment stats
- Compute read distribution across genomic features
- Adding custom features to workflow (e.g. uORFs)
- Genomic read coverage along transcripts
- Read counting
- Sample-wise correlation analysis
- Analysis of differentially expressed genes (DEGs)
- GO term enrichment analysis
- Gene-wise clustering
- Differential ribosome binding (translational efficiency)
Create the workflow
This template provides some common steps for a RIBOseq workflow. One can add, remove, modify
workflow steps by operating on the sal object.
sal <- SPRproject()
sal <- importWF(sal, file_path = "systemPipeRIBOseq.Rmd", verbose = FALSE)
Run workflow
sal <- runWF(sal)
Workflow visualization
plotWF(sal, rstudio = TRUE)
Report generation
sal <- renderReport(sal)
sal <- renderLogs(sal)
Funding
This project is funded by NSF award ABI-1661152.
Version information
Note: the most recent version of this tutorial can be found here.
sessionInfo()
## R version 4.3.0 (2023-04-21)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Debian GNU/Linux 11 (bullseye)
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: America/Los_Angeles
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] magrittr_2.0.3 batchtools_0.9.17
## [3] ape_5.7-1 ggplot2_3.4.2
## [5] systemPipeR_2.6.0 ShortRead_1.58.0
## [7] GenomicAlignments_1.36.0 SummarizedExperiment_1.30.0
## [9] Biobase_2.60.0 MatrixGenerics_1.12.0
## [11] matrixStats_0.63.0 BiocParallel_1.34.0
## [13] Rsamtools_2.16.0 Biostrings_2.68.0
## [15] XVector_0.40.0 GenomicRanges_1.52.0
## [17] GenomeInfoDb_1.36.0 IRanges_2.34.0
## [19] S4Vectors_0.38.0 BiocGenerics_0.46.0
## [21] BiocStyle_2.28.0
##
## loaded via a namespace (and not attached):
## [1] tidyselect_1.2.0 dplyr_1.1.2 bitops_1.0-7
## [4] fastmap_1.1.1 RCurl_1.98-1.12 blogdown_1.16
## [7] digest_0.6.31 base64url_1.4 lifecycle_1.0.3
## [10] compiler_4.3.0 rlang_1.1.1 sass_0.4.5
## [13] progress_1.2.2 tools_4.3.0 utf8_1.2.3
## [16] yaml_2.3.7 data.table_1.14.8 systemPipeRdata_2.4.0
## [19] knitr_1.42 prettyunits_1.1.1 brew_1.0-8
## [22] htmlwidgets_1.6.2 interp_1.1-4 DelayedArray_0.25.0
## [25] RColorBrewer_1.1-3 withr_2.5.0 hwriter_1.3.2.1
## [28] grid_4.3.0 fansi_1.0.4 latticeExtra_0.6-30
## [31] colorspace_2.1-0 scales_1.2.1 cli_3.6.1
## [34] rmarkdown_2.21 crayon_1.5.2 generics_0.1.3
## [37] remotes_2.4.2 cachem_1.0.7 stringr_1.5.0
## [40] zlibbioc_1.46.0 parallel_4.3.0 formatR_1.14
## [43] BiocManager_1.30.20 vctrs_0.6.2 Matrix_1.5-4
## [46] jsonlite_1.8.4 bookdown_0.33 hms_1.1.3
## [49] jpeg_0.1-10 jquerylib_0.1.4 glue_1.6.2
## [52] codetools_0.2-19 stringi_1.7.12 gtable_0.3.3
## [55] deldir_1.0-6 munsell_0.5.0 tibble_3.2.1
## [58] pillar_1.9.0 rappdirs_0.3.3 htmltools_0.5.5
## [61] GenomeInfoDbData_1.2.10 R6_2.5.1 evaluate_0.20
## [64] lattice_0.21-8 png_0.1-8 backports_1.4.1
## [67] bslib_0.4.2 Rcpp_1.0.10 nlme_3.1-162
## [70] checkmate_2.2.0 xfun_0.39 pkgconfig_2.0.3
References
Amstutz, Peter, Michael R Crusoe, Nebojša Tijanić, Brad Chapman, John Chilton, Michael Heuer, Andrey Kartashov, et al. 2016. “Common Workflow Language, V1.0,” July. https://doi.org/10.6084/m9.figshare.3115156.v2.
H Backman, Tyler W, and Thomas Girke. 2016. “systemPipeR: NGS workflow and report generation environment.” BMC Bioinformatics 17 (1): 388. https://doi.org/10.1186/s12859-016-1241-0.
Howard, Brian E, Qiwen Hu, Ahmet Can Babaoglu, Manan Chandra, Monica Borghi, Xiaoping Tan, Luyan He, et al. 2013. “High-Throughput RNA Sequencing of Pseudomonas-Infected Arabidopsis Reveals Hidden Transcriptome Complexity and Novel Splice Variants.” PLoS One 8 (10): e74183. https://doi.org/10.1371/journal.pone.0074183.
Kim, Daehwan, Ben Langmead, and Steven L Salzberg. 2015. “HISAT: A Fast Spliced Aligner with Low Memory Requirements.” Nat. Methods 12 (4): 357–60.