Read quality filtering and trimming
The function preprocessReads allows to apply predefined or custom
read preprocessing functions to all FASTQ files referenced in a
SYSargs container, such as quality filtering or adaptor trimming
routines. The following example performs adaptor trimming with
the trimLRPatterns function from the Biostrings package.
After the trimming step a new targets file is generated (here
targets_trim.txt) containing the paths to the trimmed FASTQ files.
The new targets file can be used for the next workflow step with an updated
SYSargs instance, e.g. running the NGS alignments using the
trimmed FASTQ files.
args <- systemArgs(sysma="param/trim.param", mytargets="targets.txt")
preprocessReads(args=args, Fct="trimLRPatterns(Rpattern='GCCCGGGTAA', subject=fq)",
batchsize=100000, overwrite=TRUE, compress=TRUE)
writeTargetsout(x=args, file="targets_trim.txt", overwrite=TRUE)FASTQ quality report
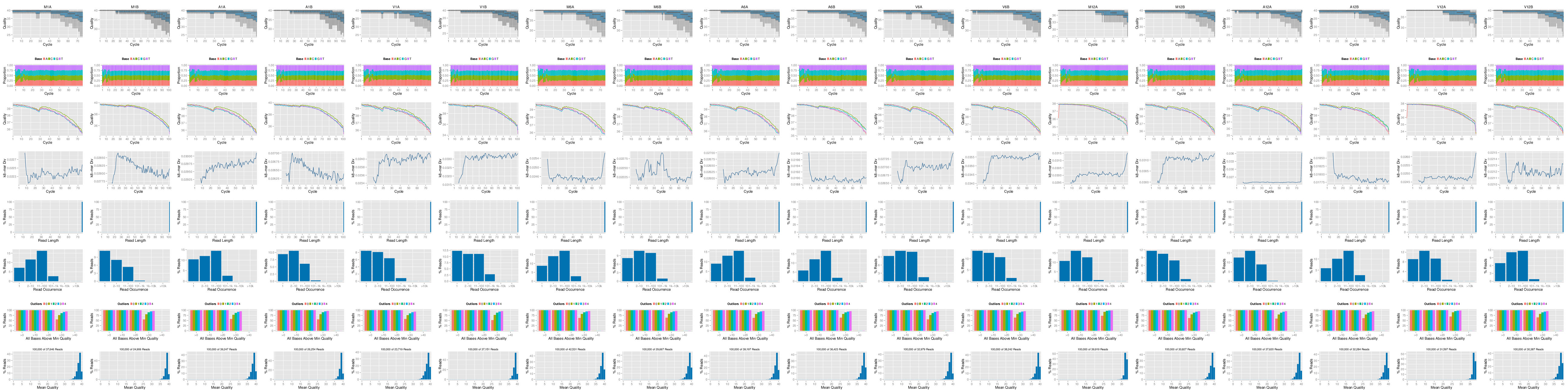
The following seeFastq and seeFastqPlot functions generate and plot a series of useful
quality statistics for a set of FASTQ files including per cycle quality box
plots, base proportions, base-level quality trends, relative k-mer
diversity, length and occurrence distribution of reads, number of reads
above quality cutoffs and mean quality distribution. The results are
written to a PDF file named fastqReport.pdf.
args <- systemArgs(sysma=NULL, mytargets="targets.txt")
fqlist <- seeFastq(fastq=infile1(args), batchsize=100000, klength=8)
png("./results/fastqReport.png", height=18, width=4*length(fqlist), units="in", res=72)
seeFastqPlot(fqlist)
dev.off()