NGS Analysis Basics
34 minute read
Overview
Sequence Analysis in R and Bioconductor
R Base
- Some basic string handling utilities. Wide spectrum of numeric data analysis tools.
Bioconductor
Bioconductor packages provide much more sophisticated string handling utilities for sequence analysis (Lawrence et al. 2013; Huber et al. 2015).
- Biostrings: general sequence analysis environment
- ShortRead: pipeline for short read data
- IRanges: low-level infrastructure for range data
- GenomicRanges: high-level infrastructure for range data
- GenomicFeatures: managing transcript centric annotations
- GenomicAlignments: handling short genomic alignments
- Rsamtools: interface to
samtools,bcftoolsandtabix - BSgenome: genome annotation data
- biomaRt: interface to BioMart annotations
- rtracklayer: Annotation imports, interface to online genome browsers
- HelloRanges: Bedtools semantics in Bioc’s Ranges infrastructure
Package Requirements
Several Bioconductor packages are required for this tutorial. To install them, execute
the following lines in the R console. Please also make sure that you have a recent R version
installed on your system. R versions 4.0.x or higher are recommended.
Please do not run this install on the HPCC unless you want to reinstall some of these packages in your own user account.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("Biostrings", "GenomicRanges", "rtracklayer", "systemPipeR", "seqLogo", "ShortRead"))
Strings in R Base
Basic String Matching and Parsing
String matching
Generate sample sequence data set
myseq <- c("ATGCAGACATAGTG", "ATGAACATAGATCC", "GTACAGATCAC")
String searching with regular expression support
myseq[grep("ATG", myseq)]
## [1] "ATGCAGACATAGTG" "ATGAACATAGATCC"
Searches myseq for first match of pattern “AT”
pos1 <- regexpr("AT", myseq)
as.numeric(pos1); attributes(pos1)$match.length # Returns position information of matches
## [1] 1 1 7
## [1] 2 2 2
Searches myseq for all matches of pattern “AT”
pos2 <- gregexpr("AT", myseq)
as.numeric(pos2[[1]]); attributes(pos2[[1]])$match.length # Returns positions of matches in first sequence
## [1] 1 9
## [1] 2 2
String substitution with regular expression support
gsub("^ATG", "atg", myseq)
## [1] "atgCAGACATAGTG" "atgAACATAGATCC" "GTACAGATCAC"
Positional parsing
nchar(myseq) # Computes length of strings
## [1] 14 14 11
substring(myseq[1], c(1,3), c(2,5)) # Positional parsing of several fragments from one string
## [1] "AT" "GCA"
substring(myseq, c(1,4,7), c(2,6,10)) # Positional parsing of many strings
## [1] "AT" "AAC" "ATCA"
Random Sequence Generation
Random DNA sequences of any length
rand <- sapply(1:100, function(x) paste(sample(c("A","T","G","C"), sample(10:20, 1), replace=TRUE), collapse=""))
rand[1:3]
## [1] "ATAACATTCCATGTCT" "CTCGACTTCGC" "AAGCCAGTTACT"
Count identical sequences
table(c(rand[1:4], rand[1]))
##
## CCAAGGGGTCACGCAAGTAA GAATACGGAAAAG GCACACCATGTT TAATCATCGAAGTT
## 1 1 2 1
Extract reads from reference
Note: this requires the Biostrings package.
library(Biostrings)
ref <- DNAString(paste(sample(c("A","T","G","C"), 100000, replace=T), collapse=""))
randstart <- sample(1:(length(ref)-15), 1000)
randreads <- Views(ref, randstart, width=15)
rand_set <- DNAStringSet(randreads)
unlist(rand_set)
## 15000-letter DNAString object
## seq: AATCGCGGCACTTAGCCAGCTTGTCTTATCGGGTATTTTGAATCTT...ACTGTCGCCAATGGGTCCAACGGATTCTACGCCACGACTCGTAACT
Sequences in Bioconductor
Important Data Objects of Biostrings
XString for single sequence
DNAString: for DNARNAString: for RNAAAString: for amino acidBString: for any string
XStringSet for many sequences
DNAStringSet: for DNARNAStringSet: for RNAAAStringSet: for amino acidBStringSet: for any string
QualityScaleXStringSet for sequences with quality data
QualityScaledDNAStringSet: for DNAQualityScaledRNAStringSet: for RNAQualityScaledAAStringSet: for amino acidQualityScaledBStringSet: for any string
Sequence Import and Export
Download the following sequences to your current working directory and then import them into R: https://ftp.ncbi.nlm.nih.gov/genomes/archive/old_genbank/Bacteria/Halobacterium_sp_uid217/AE004437.ffn
dir.create("data", showWarnings = FALSE)
# system("wget https://ftp.ncbi.nlm.nih.gov/genomes/archive/old_genbank/Bacteria/Halobacterium_sp_uid217/AE004437.ffn")
download.file("https://ftp.ncbi.nlm.nih.gov/genomes/archive/old_genbank/Bacteria/Halobacterium_sp_uid217/AE004437.ffn", "data/AE004437.ffn")
Import FASTA file with readDNAStringSet
myseq <- readDNAStringSet("data/AE004437.ffn")
myseq[1:3]
## DNAStringSet object of length 3:
## width seq names
## [1] 1206 ATGACTCGGCGGTCTCGTGTCGGTGCCGGCCTC...GTCGTCGTTGTTCGACGCTGGCGGAACCCATGA gi|12057215|gb|AE...
## [2] 666 ATGAGCATCATCGAACTCGAAGGCGTGGTCAAA...GTCAACCTCGTCGATGGGGTGTTACACACGTGA gi|12057215|gb|AE...
## [3] 1110 ATGGCGTGGCGGAACCTCGGGCGGAACCGCGTG...AACGATCCGCCCGTCGAGGCGCTCGGCGAATGA gi|12057215|gb|AE...
Subset sequences with regular expression on sequence name field
sub <- myseq[grep("99.*", names(myseq))]
length(sub)
## [1] 170
Export subsetted sequences to FASTA file
writeXStringSet(sub, file="./data/AE004437sub.ffn", width=80)
Now inspect exported sequence file AE004437sub.ffn in a text editor
Working with XString Containers
The XString stores the different types of biosequences in dedicated containers
library(Biostrings)
d <- DNAString("GCATAT-TAC")
d
## 10-letter DNAString object
## seq: GCATAT-TAC
d[1:4]
## 4-letter DNAString object
## seq: GCAT
RNA sequences
r <- RNAString("GCAUAU-UAC")
r <- RNAString(d) # Converts d to RNAString object
r
## 10-letter RNAString object
## seq: GCAUAU-UAC
Protein sequences
p <- AAString("HCWYHH")
p
## 6-letter AAString object
## seq: HCWYHH
Any type of character strings
b <- BString("I store any set of characters. Other XString objects store only the IUPAC characters.")
b
## 85-letter BString object
## seq: I store any set of characters. Other XString objects store only the IUPAC characters.
Working with XStringSet Containers
XStringSet containers allow to store many biosequences in one object
dset <- DNAStringSet(c("GCATATTAC", "AATCGATCC", "GCATATTAC"))
names(dset) <- c("seq1", "seq2", "seq3") # Assigns names
dset[1:2]
## DNAStringSet object of length 2:
## width seq names
## [1] 9 GCATATTAC seq1
## [2] 9 AATCGATCC seq2
Important utilities for XStringSet containers
width(dset) # Returns the length of each sequences
## [1] 9 9 9
d <- dset[[1]] # The [[ subsetting operator returns a single entry as XString object
dset2 <- c(dset, dset) # Appends/concatenates two XStringSet objects
dsetchar <- as.character(dset) # Converts XStringSet to named vector
dsetone <- unlist(dset) # Collapses many sequences to a single one stored in a DNAString container
Sequence subsetting by positions:
DNAStringSet(dset, start=c(1,2,3), end=c(4,8,5))
## DNAStringSet object of length 3:
## width seq names
## [1] 4 GCAT seq1
## [2] 7 ATCGATC seq2
## [3] 3 ATA seq3
Multiple Alignment Class
The XMultipleAlignment class stores the different types of multiple sequence alignments:
origMAlign <- readDNAMultipleAlignment(filepath = system.file("extdata",
"msx2_mRNA.aln", package = "Biostrings"), format = "clustal")
origMAlign
## DNAMultipleAlignment with 8 rows and 2343 columns
## aln names
## [1] -----TCCCGTCTCCGCAGCAAAAAAGTTTGAGTCG...TTGTCCAAACTCACAATTAAAAAAAAAAAAAAAAA gi|84452153|ref|N...
## [2] ------------------------------------...----------------------------------- gi|208431713|ref|...
## [3] ------------------------------------...----------------------------------- gi|118601823|ref|...
## [4] ----------------------AAAAGTTGGAGTCT...----------------------------------- gi|114326503|ref|...
## [5] ------------------------------------...----------------------------------- gi|119220589|ref|...
## [6] ------------------------------------...----------------------------------- gi|148540149|ref|...
## [7] --------------CGGCTCCGCAGCGCCTCACTCG...----------------------------------- gi|45383056|ref|N...
## [8] GGGGGAGACTTCAGAAGTTGTTGTCCTCTCCGCTGA...----------------------------------- gi|213515133|ref|...
Basic Sequence Manipulations
Reverse and Complement
randset <- DNAStringSet(rand)
complement(randset[1:2])
## DNAStringSet object of length 2:
## width seq
## [1] 12 CGTGTGGTACAA
## [2] 20 GGTTCCCCAGTGCGTTCATT
reverse(randset[1:2])
## DNAStringSet object of length 2:
## width seq
## [1] 12 TTGTACCACACG
## [2] 20 AATGAACGCACTGGGGAACC
reverseComplement(randset[1:2])
## DNAStringSet object of length 2:
## width seq
## [1] 12 AACATGGTGTGC
## [2] 20 TTACTTGCGTGACCCCTTGG
Translate DNA into Protein
translate(randset[1:2])
## Warning in .Call2("DNAStringSet_translate", x, skip_code, dna_codes[codon_alphabet], : in 'x[[2]]':
## last 2 bases were ignored
## AAStringSet object of length 2:
## width seq
## [1] 4 AHHV
## [2] 6 PRGHAS
Pattern Matching
Pattern matching with mismatches
Find pattern matches in reference
myseq1 <- readDNAStringSet("./data/AE004437.ffn")
mypos <- matchPattern("ATGGTG", myseq1[[1]], max.mismatch=1)
Count only the corresponding matches
countPattern("ATGGCT", myseq1[[1]], max.mismatch=1)
## [1] 3
Count matches in many sequences
vcountPattern("ATGGCT", myseq1, max.mismatch=1)[1:20]
## [1] 3 0 5 4 1 2 2 1 4 3 0 0 1 2 0 1 4 0 0 1
Results shown in DNAStringSet object
tmp <- c(DNAStringSet("ATGGTG"), DNAStringSet(mypos))
Return a consensus matrix for query and hits
consensusMatrix(tmp)[1:4,]
## [,1] [,2] [,3] [,4] [,5] [,6]
## A 3 0 0 0 0 0
## C 1 1 0 0 0 0
## G 0 0 4 4 1 4
## T 0 3 0 0 3 0
Find all pattern matches in reference
myvpos <- vmatchPattern("ATGGCT", myseq1, max.mismatch=1)
myvpos # The results are stored as MIndex object.
## MIndex object of length 2058
## $`gi|12057215|gb|AE004437.1|:248-1453 Halobacterium sp. NRC-1, complete genome`
## IRanges object with 3 ranges and 0 metadata columns:
## start end width
## <integer> <integer> <integer>
## [1] 1 6 6
## [2] 383 388 6
## [3] 928 933 6
##
## $`gi|12057215|gb|AE004437.1|:1450-2115 Halobacterium sp. NRC-1, complete genome`
## IRanges object with 0 ranges and 0 metadata columns:
## start end width
## <integer> <integer> <integer>
##
## $`gi|12057215|gb|AE004437.1|:2145-3254 Halobacterium sp. NRC-1, complete genome`
## IRanges object with 5 ranges and 0 metadata columns:
## start end width
## <integer> <integer> <integer>
## [1] 1 6 6
## [2] 94 99 6
## [3] 221 226 6
## [4] 535 540 6
## [5] 601 606 6
##
## ...
## <2055 more elements>
Views(myseq1[[1]], start(myvpos[[1]]), end(myvpos[[1]])) # Retrieves the result for single entry
## Views on a 1206-letter DNAString subject
## subject: ATGACTCGGCGGTCTCGTGTCGGTGCCGGCCTCGCAGCCATTGT...TTGCGATCGTCGTCGTCGTTGTTCGACGCTGGCGGAACCCATGA
## views:
## start end width
## [1] 1 6 6 [ATGACT]
## [2] 383 388 6 [ATGGCA]
## [3] 928 933 6 [ATGACT]
Return all matches
sapply(names(myseq1), function(x)
as.character(Views(myseq1[[x]], start(myvpos[[x]]), end(myvpos[[x]]))))[1:4]
Pattern matching with regular expression support
myseq <- DNAStringSet(c("ATGCAGACATAGTG", "ATGAACATAGATCC", "GTACAGATCAC"))
myseq[grep("^ATG", myseq, perl=TRUE)] # String searching with regular expression support
## DNAStringSet object of length 2:
## width seq
## [1] 14 ATGCAGACATAGTG
## [2] 14 ATGAACATAGATCC
pos1 <- regexpr("AT", myseq) # Searches 'myseq' for first match of pattern "AT"
as.numeric(pos1); attributes(pos1)$match.length # Returns position information of matches
## [1] 1 1 7
## [1] 2 2 2
pos2 <- gregexpr("AT", myseq) # Searches 'myseq' for all matches of pattern "AT"
as.numeric(pos2[[1]]); attributes(pos2[[1]])$match.length # Match positions in first sequence
## [1] 1 9
## [1] 2 2
DNAStringSet(gsub("^ATG", "NNN", myseq)) # String substitution with regular expression support
## DNAStringSet object of length 3:
## width seq
## [1] 14 NNNCAGACATAGTG
## [2] 14 NNNAACATAGATCC
## [3] 11 GTACAGATCAC
PWM Viewing and Searching
Plot with seqLogo
library(seqLogo)
pwm <- PWM(DNAStringSet(c("GCT", "GGT", "GCA")))
pwm
## [,1] [,2] [,3]
## A 0.0000000 0.0000000 0.2312611
## C 0.0000000 0.3157205 0.0000000
## G 0.3685591 0.2312611 0.0000000
## T 0.0000000 0.0000000 0.3157205
seqLogo(t(t(pwm) * 1/colSums(pwm)))
Plot with ggseqlogo
The ggseqlogo package (manual)
provides many customization options for plotting sequence logos. It also supports
various alphabets including sequence logos for amino acid sequences.
library(ggplot2); library(ggseqlogo)
pwm <- PWM(DNAStringSet(c("GCT", "GGT", "GCA")))
ggseqlogo(pwm)
## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
Search sequence for PWM matches with score better than min.score
chr <- DNAString("AAAGCTAAAGGTAAAGCAAAA")
matchPWM(pwm, chr, min.score=0.9)
## Views on a 21-letter DNAString subject
## subject: AAAGCTAAAGGTAAAGCAAAA
## views:
## start end width
## [1] 4 6 3 [GCT]
## [2] 10 12 3 [GGT]
## [3] 16 18 3 [GCA]
NGS Sequences
Sequence and Quality Data: FASTQ Format
Four lines per sequence:
- ID
- Sequence
- ID
- Base call qualities (Phred scores) as ASCII characters
The following gives an example of 3 Illumina reads in a FASTQ file. The numbers at the beginning of each line are not part of the FASTQ format. They have been added solely for illustration purposes.
1. @SRR038845.3 HWI-EAS038:6:1:0:1938 length=36
2. CAACGAGTTCACACCTTGGCCGACAGGCCCGGGTAA
3. +SRR038845.3 HWI-EAS038:6:1:0:1938 length=36
4. BA@7>B=>:>>7@7@>>9=BAA?;>52;>:9=8.=A
1. @SRR038845.41 HWI-EAS038:6:1:0:1474 length=36
2. CCAATGATTTTTTTCCGTGTTTCAGAATACGGTTAA
3. +SRR038845.41 HWI-EAS038:6:1:0:1474 length=36
4. BCCBA@BB@BBBBAB@B9B@=BABA@A:@693:@B=
1. @SRR038845.53 HWI-EAS038:6:1:1:360 length=36
2. GTTCAAAAAGAACTAAATTGTGTCAATAGAAAACTC
3. +SRR038845.53 HWI-EAS038:6:1:1:360 length=36
4. BBCBBBBBB@@BAB?BBBBCBC>BBBAA8>BBBAA@
Sequence and Quality Data: QualityScaleXStringSet
Phred quality scores are integers from 0-50 that are
stored as ASCII characters after adding 33. The basic R functions rawToChar and
charToRaw can be used to interconvert among their representations.
Phred score interconversion
phred <- 1:9
phreda <- paste(sapply(as.raw((phred)+33), rawToChar), collapse="")
phreda
## [1] "\"#$%&'()*"
as.integer(charToRaw(phreda))-33
## [1] 1 2 3 4 5 6 7 8 9
Construct QualityScaledDNAStringSet from scratch
dset <- DNAStringSet(sapply(1:100, function(x) paste(sample(c("A","T","G","C"), 20, replace=T), collapse=""))) # Creates random sample sequence.
myqlist <- lapply(1:100, function(x) sample(1:40, 20, replace=T)) # Creates random Phred score list.
myqual <- sapply(myqlist, function(x) toString(PhredQuality(x))) # Converts integer scores into ASCII characters.
myqual <- PhredQuality(myqual) # Converts to a PhredQuality object.
dsetq1 <- QualityScaledDNAStringSet(dset, myqual) # Combines DNAStringSet and quality data in QualityScaledDNAStringSet object.
dsetq1[1:2]
## A QualityScaledDNAStringSet instance containing:
##
## DNAStringSet object of length 2:
## width seq
## [1] 20 TAAAGGGCCCTACGCCTTGC
## [2] 20 TGCCTACACACAGTTAGTAC
##
## PhredQuality object of length 2:
## width seq
## [1] 20 DE4<,-D;7:E94G724C1E
## [2] 20 H#)I"/+2)3>/I@?9'8/)
Processing FASTQ Files with ShortRead
The following explains the basic usage of ShortReadQ objects. To make the sample code work,
download and unzip this file to your current working directory.
The following code performs the download for you.
library(ShortRead)
download.file("https://cluster.hpcc.ucr.edu/~tgirke/HTML_Presentations/Manuals/testdata/samplefastq/data.zip", "data.zip")
unzip("data.zip")
Important utilities for accessing FASTQ files
fastq <- list.files("data", "*.fastq$"); fastq <- paste("data/", fastq, sep="")
names(fastq) <- paste("flowcell6_lane", 1:length(fastq), sep="_")
(fq <- readFastq(fastq[1])) # Imports first FASTQ file
## class: ShortReadQ
## length: 1000 reads; width: 36 cycles
countLines(dirPath="./data", pattern=".fastq$")/4 # Counts numbers of reads in FASTQ files
## SRR038845.fastq SRR038846.fastq SRR038848.fastq SRR038850.fastq
## 1000 1000 1000 1000
id(fq)[1] # Returns ID field
## BStringSet object of length 1:
## width seq
## [1] 43 SRR038845.3 HWI-EAS038:6:1:0:1938 length=36
sread(fq)[1] # Returns sequence
## DNAStringSet object of length 1:
## width seq
## [1] 36 CAACGAGTTCACACCTTGGCCGACAGGCCCGGGTAA
quality(fq)[1] # Returns Phred scores
## class: FastqQuality
## quality:
## BStringSet object of length 1:
## width seq
## [1] 36 BA@7>B=>:>>7@7@>>9=BAA?;>52;>:9=8.=A
as(quality(fq), "matrix")[1:4,1:12] # Coerces Phred scores to numeric matrix
## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12]
## [1,] 33 32 31 22 29 33 28 29 25 29 29 22
## [2,] 33 34 34 33 32 31 33 33 31 33 33 33
## [3,] 33 33 34 33 33 33 33 33 33 31 31 33
## [4,] 33 33 33 33 31 33 28 31 28 32 33 33
ShortReadQ(sread=sread(fq), quality=quality(fq), id=id(fq)) # Constructs a ShortReadQ from components
## class: ShortReadQ
## length: 1000 reads; width: 36 cycles
FASTQ Quality Reports
Using systemPipeR
The following seeFastq and seeFastqPlot functions generate and plot a series of useful quality statistics for a set of FASTQ files.
library(systemPipeR)
fqlist <- seeFastq(fastq=fastq, batchsize=800, klength=8) # For real data set batchsize to at least 10^5
seeFastqPlot(fqlist)
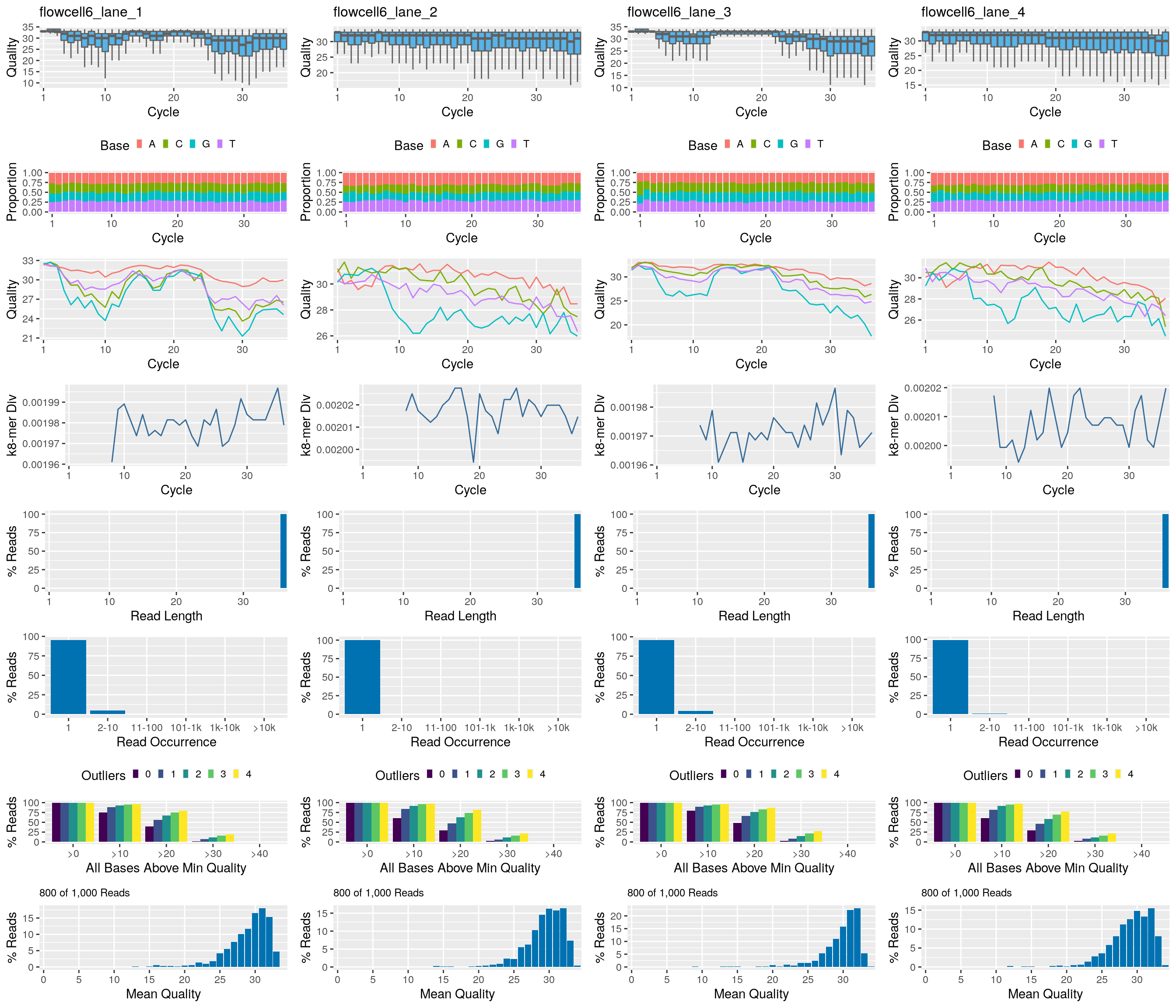
Handles many samples in one PDF file. For more details see here
Using ShortRead
The ShortRead package contains several FASTQ quality reporting functions.
sp <- SolexaPath(system.file('extdata', package='ShortRead'))
fl <- file.path(analysisPath(sp), "s_1_sequence.txt")
fls <- c(fl, fl)
coll <- QACollate(QAFastqSource(fls), QAReadQuality(), QAAdapterContamination(),
QANucleotideUse(), QAQualityUse(), QASequenceUse(), QAFrequentSequence(n=10),
QANucleotideByCycle(), QAQualityByCycle())
x <- qa2(coll, verbose=TRUE)
res <- report(x)
if(interactive())
browseURL(res)
Filtering and Trimming FASTQ Files with ShortRead
Adaptor trimming
fqtrim <- trimLRPatterns(Rpattern="GCCCGGGTAA", subject=fq)
sread(fq)[1:2] # Before trimming
## DNAStringSet object of length 2:
## width seq
## [1] 36 CAACGAGTTCACACCTTGGCCGACAGGCCCGGGTAA
## [2] 36 CCAATGATTTTTTTCCGTGTTTCAGAATACGGTTAA
sread(fqtrim)[1:2] # After trimming
## DNAStringSet object of length 2:
## width seq
## [1] 26 CAACGAGTTCACACCTTGGCCGACAG
## [2] 36 CCAATGATTTTTTTCCGTGTTTCAGAATACGGTTAA
Read counting and duplicate removal
tables(fq)$distribution # Counts read occurences
## nOccurrences nReads
## 1 1 948
## 2 2 26
sum(srduplicated(fq)) # Identifies duplicated reads
## [1] 26
fq[!srduplicated(fq)]
## class: ShortReadQ
## length: 974 reads; width: 36 cycles
Trimming low quality tails
cutoff <- 30
cutoff <- rawToChar(as.raw(cutoff+33))
sread(trimTails(fq, k=2, a=cutoff, successive=FALSE))[1:2]
## DNAStringSet object of length 2:
## width seq
## [1] 4 CAAC
## [2] 20 CCAATGATTTTTTTCCGTGT
Removal of reads with Phred scores below a threshold value
cutoff <- 30
qcount <- rowSums(as(quality(fq), "matrix") <= 20)
fq[qcount == 0] # Number of reads where all Phred scores >= 20
## class: ShortReadQ
## length: 349 reads; width: 36 cycles
Removal of reads with x Ns and/or low complexity segments
filter1 <- nFilter(threshold=1) # Keeps only reads without Ns
filter2 <- polynFilter(threshold=20, nuc=c("A","T","G","C")) # Removes reads with nucleotide bias, >=20 of any base
filter <- compose(filter1, filter2)
fq[filter(fq)]
## class: ShortReadQ
## length: 989 reads; width: 36 cycles
Memory Efficient FASTQ Processing
Streaming through FASTQ files with FastqStreamer and random sampling reads with FastqSampler
fq <- yield(FastqStreamer(fastq[1], 50)) # Imports first 50 reads
fq <- yield(FastqSampler(fastq[1], 50)) # Random samples 50 reads
Streaming through a FASTQ file while applying filtering/trimming functions and writing the results to a new file
here SRR038845.fastq_sub in data directory.
f <- FastqStreamer(fastq[1], 50)
while(length(fq <- yield(f))) {
fqsub <- fq[grepl("^TT", sread(fq))]
writeFastq(fqsub, paste(fastq[1], "sub", sep="_"), mode="a", compress=FALSE)
}
close(f)
Range Operations
Important Data Objects for Range Operations
IRanges: stores range data only (IRanges library)GRanges: stores ranges and annotations (GenomicRanges library)GRangesList: list version of GRanges container (GenomicRanges library)
Range Data Are Stored in IRanges and GRanges Containers
Construct GRanges Object
The following works with information that relates to GFF/GTF file format. The defintions of the GFF/GTF file format are here.
library(GenomicRanges); library(rtracklayer)
gr <- GRanges(seqnames = Rle(c("chr1", "chr2", "chr1", "chr3"), c(1, 3, 2, 4)), ranges = IRanges(1:10, end = 7:16, names = head(letters, 10)), strand = Rle(strand(c("-", "+", "*", "+", "-")), c(1, 2, 2, 3, 2)), score = 1:10, GC = seq(1, 0, length = 10)) # Example of creating a GRanges object with its constructor function.
Import GFF into GRanges Object
gff <- import.gff("https://cluster.hpcc.ucr.edu/~tgirke/Documents/R_BioCond/Samples/gff3.gff") # Imports a simplified GFF3 genome annotation file.
seqlengths(gff) <- end(ranges(gff[which(values(gff)[,"type"]=="chromosome"),]))
names(gff) <- 1:length(gff) # Assigns names to corresponding slot
gff[1:4,]
## GRanges object with 4 ranges and 10 metadata columns:
## seqnames ranges strand | source type score phase ID
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer> <character>
## 1 Chr1 1-30427671 + | TAIR10 chromosome NA <NA> Chr1
## 2 Chr1 3631-5899 + | TAIR10 gene NA <NA> AT1G01010
## 3 Chr1 3631-5899 + | TAIR10 mRNA NA <NA> AT1G01010.1
## 4 Chr1 3760-5630 + | TAIR10 protein NA <NA> AT1G01010.1-Protein
## Name Note Parent Index Derives_from
## <character> <CharacterList> <CharacterList> <character> <character>
## 1 Chr1 <NA> <NA>
## 2 AT1G01010 protein_coding_gene <NA> <NA>
## 3 AT1G01010.1 AT1G01010 1 <NA>
## 4 AT1G01010.1 <NA> AT1G01010.1
## -------
## seqinfo: 7 sequences from an unspecified genome
seqinfo(gff)
## Seqinfo object with 7 sequences from an unspecified genome:
## seqnames seqlengths isCircular genome
## Chr1 30427671 NA <NA>
## Chr2 19698289 NA <NA>
## Chr3 23459830 NA <NA>
## Chr4 18585056 NA <NA>
## Chr5 26975502 NA <NA>
## ChrC 154478 NA <NA>
## ChrM 366924 NA <NA>
Coerce GRanges object to data.frame
as.data.frame(gff)[1:4, 1:7]
## seqnames start end width strand source type
## 1 Chr1 1 30427671 30427671 + TAIR10 chromosome
## 2 Chr1 3631 5899 2269 + TAIR10 gene
## 3 Chr1 3631 5899 2269 + TAIR10 mRNA
## 4 Chr1 3760 5630 1871 + TAIR10 protein
Utilities for Range Containers
Accessor and subsetting methods for GRanges objects
Subsetting and replacement
gff[1:4]
## GRanges object with 4 ranges and 10 metadata columns:
## seqnames ranges strand | source type score phase ID
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer> <character>
## 1 Chr1 1-30427671 + | TAIR10 chromosome NA <NA> Chr1
## 2 Chr1 3631-5899 + | TAIR10 gene NA <NA> AT1G01010
## 3 Chr1 3631-5899 + | TAIR10 mRNA NA <NA> AT1G01010.1
## 4 Chr1 3760-5630 + | TAIR10 protein NA <NA> AT1G01010.1-Protein
## Name Note Parent Index Derives_from
## <character> <CharacterList> <CharacterList> <character> <character>
## 1 Chr1 <NA> <NA>
## 2 AT1G01010 protein_coding_gene <NA> <NA>
## 3 AT1G01010.1 AT1G01010 1 <NA>
## 4 AT1G01010.1 <NA> AT1G01010.1
## -------
## seqinfo: 7 sequences from an unspecified genome
gff[1:4, c("type", "ID")]
## GRanges object with 4 ranges and 2 metadata columns:
## seqnames ranges strand | type ID
## <Rle> <IRanges> <Rle> | <factor> <character>
## 1 Chr1 1-30427671 + | chromosome Chr1
## 2 Chr1 3631-5899 + | gene AT1G01010
## 3 Chr1 3631-5899 + | mRNA AT1G01010.1
## 4 Chr1 3760-5630 + | protein AT1G01010.1-Protein
## -------
## seqinfo: 7 sequences from an unspecified genome
gff[2] <- gff[3]
GRanges objects can be concatenated with the c function
c(gff[1:2], gff[401:402])
## GRanges object with 4 ranges and 10 metadata columns:
## seqnames ranges strand | source type score phase ID
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer> <character>
## 1 Chr1 1-30427671 + | TAIR10 chromosome NA <NA> Chr1
## 2 Chr1 3631-5899 + | TAIR10 mRNA NA <NA> AT1G01010.1
## 401 Chr5 5516-5769 - | TAIR10 protein NA <NA> AT5G01015.2-Protein
## 402 Chr5 5770-5801 - | TAIR10 five_prime_UTR NA <NA> <NA>
## Name Note Parent Index Derives_from
## <character> <CharacterList> <CharacterList> <character> <character>
## 1 Chr1 <NA> <NA>
## 2 AT1G01010.1 AT1G01010 1 <NA>
## 401 AT5G01015.2 <NA> AT5G01015.2
## 402 <NA> AT5G01015.2 <NA> <NA>
## -------
## seqinfo: 7 sequences from an unspecified genome
Acessor functions
seqnames(gff)
## factor-Rle of length 449 with 7 runs
## Lengths: 72 22 38 118 172 13 14
## Values : Chr1 Chr2 Chr3 Chr4 Chr5 ChrC ChrM
## Levels(7): Chr1 Chr2 Chr3 Chr4 Chr5 ChrC ChrM
ranges(gff)
## IRanges object with 449 ranges and 0 metadata columns:
## start end width
## <integer> <integer> <integer>
## 1 1 30427671 30427671
## 2 3631 5899 2269
## 3 3631 5899 2269
## 4 3760 5630 1871
## 5 3631 3913 283
## ... ... ... ...
## 445 11918 12241 324
## 446 11918 12241 324
## 447 11918 12241 324
## 448 11918 12241 324
## 449 11918 12241 324
strand(gff)
## factor-Rle of length 449 with 13 runs
## Lengths: 18 54 28 21 12 117 1 171 1 12 1 8 5
## Values : + - + - + - + - + - + - +
## Levels(3): + - *
seqlengths(gff)
## Chr1 Chr2 Chr3 Chr4 Chr5 ChrC ChrM
## 30427671 19698289 23459830 18585056 26975502 154478 366924
start(gff[1:4])
## [1] 1 3631 3631 3760
end(gff[1:4])
## [1] 30427671 5899 5899 5630
width(gff[1:4])
## [1] 30427671 2269 2269 1871
Accessing metadata component
values(gff) # or elementMetadata(gff)
## DataFrame with 449 rows and 10 columns
## source type score phase ID Name Note
## <factor> <factor> <numeric> <integer> <character> <character> <CharacterList>
## 1 TAIR10 chromosome NA NA Chr1 Chr1
## 2 TAIR10 mRNA NA NA AT1G01010.1 AT1G01010.1
## 3 TAIR10 mRNA NA NA AT1G01010.1 AT1G01010.1
## 4 TAIR10 protein NA NA AT1G01010.1-Protein AT1G01010.1
## 5 TAIR10 exon NA NA NA NA
## ... ... ... ... ... ... ... ...
## 445 TAIR10 gene NA NA ATMG00030 ATMG00030 protein_coding_gene
## 446 TAIR10 mRNA NA NA ATMG00030.1 ATMG00030.1
## 447 TAIR10 protein NA NA ATMG00030.1-Protein ATMG00030.1
## 448 TAIR10 exon NA NA NA NA
## 449 TAIR10 CDS NA 0 NA NA
## Parent Index Derives_from
## <CharacterList> <character> <character>
## 1 NA NA
## 2 AT1G01010 1 NA
## 3 AT1G01010 1 NA
## 4 NA AT1G01010.1
## 5 AT1G01010.1 NA NA
## ... ... ... ...
## 445 NA NA
## 446 ATMG00030 1 NA
## 447 NA ATMG00030.1
## 448 ATMG00030.1 NA NA
## 449 ATMG00030.1,ATMG00030.1-Protein NA NA
values(gff)[, "type"][1:20]
## [1] chromosome mRNA mRNA protein exon five_prime_UTR
## [7] CDS exon CDS exon CDS exon
## [13] CDS exon CDS exon CDS three_prime_UTR
## [19] gene mRNA
## Levels: chromosome gene mRNA protein exon five_prime_UTR CDS three_prime_UTR rRNA tRNA
gff[values(gff)[ ,"type"] == "gene"]
## GRanges object with 21 ranges and 10 metadata columns:
## seqnames ranges strand | source type score phase ID Name
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer> <character> <character>
## 19 Chr1 5928-8737 - | TAIR10 gene NA <NA> AT1G01020 AT1G01020
## 64 Chr1 11649-13714 - | TAIR10 gene NA <NA> AT1G01030 AT1G01030
## 74 Chr2 1025-2810 + | TAIR10 gene NA <NA> AT2G01008 AT2G01008
## 84 Chr2 3706-5513 + | TAIR10 gene NA <NA> AT2G01010 AT2G01010
## 87 Chr2 5782-5945 + | TAIR10 gene NA <NA> AT2G01020 AT2G01020
## ... ... ... ... . ... ... ... ... ... ...
## 427 ChrC 383-1444 - | TAIR10 gene NA <NA> ATCG00020 ATCG00020
## 432 ChrC 1717-4347 - | TAIR10 gene NA <NA> ATCG00030 ATCG00030
## 437 ChrM 273-734 - | TAIR10 gene NA <NA> ATMG00010 ATMG00010
## 442 ChrM 8848-11415 - | TAIR10 gene NA <NA> ATMG00020 ATMG00020
## 445 ChrM 11918-12241 + | TAIR10 gene NA <NA> ATMG00030 ATMG00030
## Note Parent Index Derives_from
## <CharacterList> <CharacterList> <character> <character>
## 19 protein_coding_gene <NA> <NA>
## 64 protein_coding_gene <NA> <NA>
## 74 protein_coding_gene <NA> <NA>
## 84 rRNA <NA> <NA>
## 87 rRNA <NA> <NA>
## ... ... ... ... ...
## 427 protein_coding_gene <NA> <NA>
## 432 tRNA <NA> <NA>
## 437 protein_coding_gene <NA> <NA>
## 442 rRNA <NA> <NA>
## 445 protein_coding_gene <NA> <NA>
## -------
## seqinfo: 7 sequences from an unspecified genome
Useful utilities for GRanges objects
Remove chromosome ranges
gff <- gff[values(gff)$type != "chromosome"]
Erase the strand information
strand(gff) <- "*"
Collapses overlapping ranges to continuous ranges. To ignore strand information, include argument ignore.strand=TRUE.
reduce(gff)
## GRanges object with 22 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] Chr1 3631-5899 *
## [2] Chr1 5928-8737 *
## [3] Chr1 11649-13714 *
## [4] Chr2 1025-2810 *
## [5] Chr2 3706-5513 *
## ... ... ... ...
## [18] ChrC 383-1444 *
## [19] ChrC 1717-4347 *
## [20] ChrM 273-734 *
## [21] ChrM 8848-11415 *
## [22] ChrM 11918-12241 *
## -------
## seqinfo: 7 sequences from an unspecified genome
Return uncovered regions
gaps(gff)
## GRanges object with 43 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] Chr1 1-30427671 +
## [2] Chr1 1-30427671 -
## [3] Chr1 1-3630 *
## [4] Chr1 5900-5927 *
## [5] Chr1 8738-11648 *
## ... ... ... ...
## [39] ChrM 1-366924 -
## [40] ChrM 1-272 *
## [41] ChrM 735-8847 *
## [42] ChrM 11416-11917 *
## [43] ChrM 12242-366924 *
## -------
## seqinfo: 7 sequences from an unspecified genome
More intuitive way to get uncovered regions
setdiff(as(seqinfo(gff), "GRanges"), gff)
## GRanges object with 29 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] Chr1 1-3630 *
## [2] Chr1 5900-5927 *
## [3] Chr1 8738-11648 *
## [4] Chr1 13715-30427671 *
## [5] Chr2 1-1024 *
## ... ... ... ...
## [25] ChrC 4348-154478 *
## [26] ChrM 1-272 *
## [27] ChrM 735-8847 *
## [28] ChrM 11416-11917 *
## [29] ChrM 12242-366924 *
## -------
## seqinfo: 7 sequences from an unspecified genome
Return disjoint ranges
disjoin(gff)
## GRanges object with 211 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] Chr1 3631-3759 *
## [2] Chr1 3760-3913 *
## [3] Chr1 3914-3995 *
## [4] Chr1 3996-4276 *
## [5] Chr1 4277-4485 *
## ... ... ... ...
## [207] ChrC 1752-4310 *
## [208] ChrC 4311-4347 *
## [209] ChrM 273-734 *
## [210] ChrM 8848-11415 *
## [211] ChrM 11918-12241 *
## -------
## seqinfo: 7 sequences from an unspecified genome
Returns coverage of ranges
coverage(gff)
## RleList of length 7
## $Chr1
## integer-Rle of length 30427671 with 45 runs
## Lengths: 3630 129 154 82 281 ... 233 161 380 30413957
## Values : 0 4 5 3 5 ... 4 2 4 0
##
## $Chr2
## integer-Rle of length 19698289 with 14 runs
## Lengths: 1024 248 185 53 362 ... 164 625 102 19691617
## Values : 0 5 3 5 3 ... 3 0 5 0
##
## $Chr3
## integer-Rle of length 23459830 with 29 runs
## Lengths: 1652 145 139 111 95 ... 155 148 156 23453781
## Values : 0 4 5 3 5 ... 3 5 4 0
##
## $Chr4
## integer-Rle of length 18585056 with 72 runs
## Lengths: 1179 357 1358 128 872 ... 212 114 74 18571697
## Values : 0 5 0 5 3 ... 3 5 4 0
##
## $Chr5
## integer-Rle of length 26975502 with 64 runs
## Lengths: 1222 28 28 109 72 ... 76 55 174 26967058
## Values : 0 4 7 13 16 ... 3 5 4 0
##
## ...
## <2 more elements>
Return the index pairings for overlapping ranges
findOverlaps(gff, gff[1:4])
## Hits object with 55 hits and 0 metadata columns:
## queryHits subjectHits
## <integer> <integer>
## [1] 1 1
## [2] 1 2
## [3] 1 4
## [4] 1 3
## [5] 2 1
## ... ... ...
## [51] 16 1
## [52] 16 2
## [53] 16 3
## [54] 17 1
## [55] 17 2
## -------
## queryLength: 442 / subjectLength: 4
Counts overlapping ranges
countOverlaps(gff, gff[1:4])[1:40]
## 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34
## 4 4 4 4 3 4 3 3 3 3 3 3 3 3 3 3 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
## 35 36 37 38 39 40 41
## 0 0 0 0 0 0 0
Return only overlapping ranges
subsetByOverlaps(gff, gff[1:4])
## GRanges object with 17 ranges and 10 metadata columns:
## seqnames ranges strand | source type score phase ID
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer> <character>
## 2 Chr1 3631-5899 * | TAIR10 mRNA NA <NA> AT1G01010.1
## 3 Chr1 3631-5899 * | TAIR10 mRNA NA <NA> AT1G01010.1
## 4 Chr1 3760-5630 * | TAIR10 protein NA <NA> AT1G01010.1-Protein
## 5 Chr1 3631-3913 * | TAIR10 exon NA <NA> <NA>
## 6 Chr1 3631-3759 * | TAIR10 five_prime_UTR NA <NA> <NA>
## .. ... ... ... . ... ... ... ... ...
## 14 Chr1 5174-5326 * | TAIR10 exon NA <NA> <NA>
## 15 Chr1 5174-5326 * | TAIR10 CDS NA 0 <NA>
## 16 Chr1 5439-5899 * | TAIR10 exon NA <NA> <NA>
## 17 Chr1 5439-5630 * | TAIR10 CDS NA 0 <NA>
## 18 Chr1 5631-5899 * | TAIR10 three_prime_UTR NA <NA> <NA>
## Name Note Parent Index Derives_from
## <character> <CharacterList> <CharacterList> <character> <character>
## 2 AT1G01010.1 AT1G01010 1 <NA>
## 3 AT1G01010.1 AT1G01010 1 <NA>
## 4 AT1G01010.1 <NA> AT1G01010.1
## 5 <NA> AT1G01010.1 <NA> <NA>
## 6 <NA> AT1G01010.1 <NA> <NA>
## .. ... ... ... ... ...
## 14 <NA> AT1G01010.1 <NA> <NA>
## 15 <NA> AT1G01010.1,AT1G01010.1-Protein <NA> <NA>
## 16 <NA> AT1G01010.1 <NA> <NA>
## 17 <NA> AT1G01010.1,AT1G01010.1-Protein <NA> <NA>
## 18 <NA> AT1G01010.1 <NA> <NA>
## -------
## seqinfo: 7 sequences from an unspecified genome
GRangesList Objects
sp <- split(gff, seq(along=gff)) # Stores every range in separate component of a GRangesList object
split(gff, seqnames(gff)) # Stores ranges of each chromosome in separate component.
## GRangesList object of length 7:
## $Chr1
## GRanges object with 71 ranges and 10 metadata columns:
## seqnames ranges strand | source type score phase ID
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer> <character>
## 2 Chr1 3631-5899 * | TAIR10 mRNA NA <NA> AT1G01010.1
## 3 Chr1 3631-5899 * | TAIR10 mRNA NA <NA> AT1G01010.1
## 4 Chr1 3760-5630 * | TAIR10 protein NA <NA> AT1G01010.1-Protein
## 5 Chr1 3631-3913 * | TAIR10 exon NA <NA> <NA>
## 6 Chr1 3631-3759 * | TAIR10 five_prime_UTR NA <NA> <NA>
## .. ... ... ... . ... ... ... ... ...
## 68 Chr1 13335-13714 * | TAIR10 exon NA <NA> <NA>
## 69 Chr1 12941-13173 * | TAIR10 five_prime_UTR NA <NA> <NA>
## 70 Chr1 11864-12940 * | TAIR10 CDS NA 0 <NA>
## 71 Chr1 11649-11863 * | TAIR10 three_prime_UTR NA <NA> <NA>
## 72 Chr1 11649-13173 * | TAIR10 exon NA <NA> <NA>
## Name Note Parent Index Derives_from
## <character> <CharacterList> <CharacterList> <character> <character>
## 2 AT1G01010.1 AT1G01010 1 <NA>
## 3 AT1G01010.1 AT1G01010 1 <NA>
## 4 AT1G01010.1 <NA> AT1G01010.1
## 5 <NA> AT1G01010.1 <NA> <NA>
## 6 <NA> AT1G01010.1 <NA> <NA>
## .. ... ... ... ... ...
## 68 <NA> AT1G01030.1 <NA> <NA>
## 69 <NA> AT1G01030.1 <NA> <NA>
## 70 <NA> AT1G01030.1,AT1G01030.1-Protein <NA> <NA>
## 71 <NA> AT1G01030.1 <NA> <NA>
## 72 <NA> AT1G01030.1 <NA> <NA>
## -------
## seqinfo: 7 sequences from an unspecified genome
##
## ...
## <6 more elements>
unlist(sp) # Returns data as GRanges object
## GRanges object with 442 ranges and 10 metadata columns:
## seqnames ranges strand | source type score phase
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric> <integer>
## 1.2 Chr1 3631-5899 * | TAIR10 mRNA NA <NA>
## 2.3 Chr1 3631-5899 * | TAIR10 mRNA NA <NA>
## 3.4 Chr1 3760-5630 * | TAIR10 protein NA <NA>
## 4.5 Chr1 3631-3913 * | TAIR10 exon NA <NA>
## 5.6 Chr1 3631-3759 * | TAIR10 five_prime_UTR NA <NA>
## ... ... ... ... . ... ... ... ...
## 438.445 ChrM 11918-12241 * | TAIR10 gene NA <NA>
## 439.446 ChrM 11918-12241 * | TAIR10 mRNA NA <NA>
## 440.447 ChrM 11918-12241 * | TAIR10 protein NA <NA>
## 441.448 ChrM 11918-12241 * | TAIR10 exon NA <NA>
## 442.449 ChrM 11918-12241 * | TAIR10 CDS NA 0
## ID Name Note Parent
## <character> <character> <CharacterList> <CharacterList>
## 1.2 AT1G01010.1 AT1G01010.1 AT1G01010
## 2.3 AT1G01010.1 AT1G01010.1 AT1G01010
## 3.4 AT1G01010.1-Protein AT1G01010.1
## 4.5 <NA> <NA> AT1G01010.1
## 5.6 <NA> <NA> AT1G01010.1
## ... ... ... ... ...
## 438.445 ATMG00030 ATMG00030 protein_coding_gene
## 439.446 ATMG00030.1 ATMG00030.1 ATMG00030
## 440.447 ATMG00030.1-Protein ATMG00030.1
## 441.448 <NA> <NA> ATMG00030.1
## 442.449 <NA> <NA> ATMG00030.1,ATMG00030.1-Protein
## Index Derives_from
## <character> <character>
## 1.2 1 <NA>
## 2.3 1 <NA>
## 3.4 <NA> AT1G01010.1
## 4.5 <NA> <NA>
## 5.6 <NA> <NA>
## ... ... ...
## 438.445 <NA> <NA>
## 439.446 1 <NA>
## 440.447 <NA> ATMG00030.1
## 441.448 <NA> <NA>
## 442.449 <NA> <NA>
## -------
## seqinfo: 7 sequences from an unspecified genome
sp[1:4, "type"] # Subsetting of GRangesList objects is similar to GRanges objects.
## GRangesList object of length 4:
## $`1`
## GRanges object with 1 range and 1 metadata column:
## seqnames ranges strand | type
## <Rle> <IRanges> <Rle> | <factor>
## 2 Chr1 3631-5899 * | mRNA
## -------
## seqinfo: 7 sequences from an unspecified genome
##
## $`2`
## GRanges object with 1 range and 1 metadata column:
## seqnames ranges strand | type
## <Rle> <IRanges> <Rle> | <factor>
## 3 Chr1 3631-5899 * | mRNA
## -------
## seqinfo: 7 sequences from an unspecified genome
##
## $`3`
## GRanges object with 1 range and 1 metadata column:
## seqnames ranges strand | type
## <Rle> <IRanges> <Rle> | <factor>
## 4 Chr1 3760-5630 * | protein
## -------
## seqinfo: 7 sequences from an unspecified genome
##
## $`4`
## GRanges object with 1 range and 1 metadata column:
## seqnames ranges strand | type
## <Rle> <IRanges> <Rle> | <factor>
## 5 Chr1 3631-3913 * | exon
## -------
## seqinfo: 7 sequences from an unspecified genome
lapply(sp[1:4], length) # Looping over GRangesList objects similar to lists
## $`1`
## [1] 1
##
## $`2`
## [1] 1
##
## $`3`
## [1] 1
##
## $`4`
## [1] 1
Transcript Ranges
Storing annotation ranges in TranscriptDb databases makes many operations more robust and convenient.
library(GenomicFeatures)
download.file("https://cluster.hpcc.ucr.edu/~tgirke/Documents/R_BioCond/Samples/gff3.gff", "data/gff3.gff")
txdb <- makeTxDbFromGFF(file="data/gff3.gff", format="gff", dataSource="TAIR", organism="Arabidopsis thaliana")
## Warning in call_fun_in_txdbmaker("makeTxDbFromGFF", ...): makeTxDbFromGFF() has moved to the txdbmaker package. Please call
## txdbmaker::makeTxDbFromGFF() to get rid of this warning.
## Warning in .extract_exons_from_GRanges(cds_IDX, gr, mcols0, tx_IDX, feature = "cds", : 163 CDS couldn't be linked to a transcript so were dropped (showing only the first 6):
## seqid start end strand ID Name Parent Parent_type
## 1 Chr1 3760 3913 + <NA> <NA> AT1G01010.1-Protein <NA>
## 2 Chr1 3996 4276 + <NA> <NA> AT1G01010.1-Protein <NA>
## 3 Chr1 4486 4605 + <NA> <NA> AT1G01010.1-Protein <NA>
## 4 Chr1 4706 5095 + <NA> <NA> AT1G01010.1-Protein <NA>
## 5 Chr1 5174 5326 + <NA> <NA> AT1G01010.1-Protein <NA>
## 6 Chr1 5439 5630 + <NA> <NA> AT1G01010.1-Protein <NA>
saveDb(txdb, file="./data/TAIR10.sqlite")
## TxDb object:
## # Db type: TxDb
## # Supporting package: GenomicFeatures
## # Data source: TAIR
## # Organism: Arabidopsis thaliana
## # Taxonomy ID: 3702
## # miRBase build ID: NA
## # Genome: NA
## # Nb of transcripts: 28
## # Db created by: txdbmaker package from Bioconductor
## # Creation time: 2025-04-24 19:19:56 -0700 (Thu, 24 Apr 2025)
## # txdbmaker version at creation time: 1.2.1
## # RSQLite version at creation time: 2.3.9
## # DBSCHEMAVERSION: 1.2
txdb <- loadDb("./data/TAIR10.sqlite")
transcripts(txdb)
## GRanges object with 28 ranges and 2 metadata columns:
## seqnames ranges strand | tx_id tx_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] Chr1 3631-5899 + | 1 AT1G01010.1
## [2] Chr1 5928-8737 - | 2 AT1G01020.1
## [3] Chr1 6790-8737 - | 3 AT1G01020.2
## [4] Chr1 11649-13714 - | 4 AT1G01030.1
## [5] Chr2 1025-2810 + | 5 AT2G01008.1
## ... ... ... ... . ... ...
## [24] ChrC 383-1444 - | 24 ATCG00020.1
## [25] ChrC 1717-4347 - | 25 ATCG00030.1
## [26] ChrM 11918-12241 + | 26 ATMG00030.1
## [27] ChrM 273-734 - | 27 ATMG00010.1
## [28] ChrM 8848-11415 - | 28 ATMG00020.1
## -------
## seqinfo: 7 sequences (2 circular) from an unspecified genome; no seqlengths
transcriptsBy(txdb, by = "gene")
## GRangesList object of length 22:
## $AT1G01010
## GRanges object with 1 range and 2 metadata columns:
## seqnames ranges strand | tx_id tx_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] Chr1 3631-5899 + | 1 AT1G01010.1
## -------
## seqinfo: 7 sequences (2 circular) from an unspecified genome; no seqlengths
##
## $AT1G01020
## GRanges object with 2 ranges and 2 metadata columns:
## seqnames ranges strand | tx_id tx_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] Chr1 5928-8737 - | 2 AT1G01020.1
## [2] Chr1 6790-8737 - | 3 AT1G01020.2
## -------
## seqinfo: 7 sequences (2 circular) from an unspecified genome; no seqlengths
##
## $AT1G01030
## GRanges object with 1 range and 2 metadata columns:
## seqnames ranges strand | tx_id tx_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] Chr1 11649-13714 - | 4 AT1G01030.1
## -------
## seqinfo: 7 sequences (2 circular) from an unspecified genome; no seqlengths
##
## ...
## <19 more elements>
exonsBy(txdb, by = "gene")
## GRangesList object of length 22:
## $AT1G01010
## GRanges object with 6 ranges and 2 metadata columns:
## seqnames ranges strand | exon_id exon_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] Chr1 3631-3913 + | 1 <NA>
## [2] Chr1 3996-4276 + | 2 <NA>
## [3] Chr1 4486-4605 + | 3 <NA>
## [4] Chr1 4706-5095 + | 4 <NA>
## [5] Chr1 5174-5326 + | 5 <NA>
## [6] Chr1 5439-5899 + | 6 <NA>
## -------
## seqinfo: 7 sequences (2 circular) from an unspecified genome; no seqlengths
##
## $AT1G01020
## GRanges object with 12 ranges and 2 metadata columns:
## seqnames ranges strand | exon_id exon_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] Chr1 5928-6263 - | 7 <NA>
## [2] Chr1 6437-7069 - | 8 <NA>
## [3] Chr1 6790-7069 - | 9 <NA>
## [4] Chr1 7157-7232 - | 10 <NA>
## [5] Chr1 7157-7450 - | 11 <NA>
## ... ... ... ... . ... ...
## [8] Chr1 7762-7835 - | 14 <NA>
## [9] Chr1 7942-7987 - | 15 <NA>
## [10] Chr1 8236-8325 - | 16 <NA>
## [11] Chr1 8417-8464 - | 17 <NA>
## [12] Chr1 8571-8737 - | 18 <NA>
## -------
## seqinfo: 7 sequences (2 circular) from an unspecified genome; no seqlengths
##
## $AT1G01030
## GRanges object with 2 ranges and 2 metadata columns:
## seqnames ranges strand | exon_id exon_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] Chr1 11649-13173 - | 19 <NA>
## [2] Chr1 13335-13714 - | 20 <NA>
## -------
## seqinfo: 7 sequences (2 circular) from an unspecified genome; no seqlengths
##
## ...
## <19 more elements>
txdb from BioMart
Alternative sources for creating txdb databases are BioMart, Bioc annotation packages, UCSC, etc. The following shows how to create a txdb from BioMart.
library(GenomicFeatures); library("biomaRt")
txdb <- makeTxDbFromBiomart(biomart = "plants_mart", dataset = "athaliana_eg_gene", host="https://plants.ensembl.org")
The following steps are useful to find out what is availble in BioMart.
listMarts() # Lists BioMart databases
listMarts(host="plants.ensembl.org")
mymart <- useMart("plants_mart", host="plants.ensembl.org") # Select one, here plants_mart
listDatasets(mymart) # List datasets available in the selected BioMart database
mymart <- useMart("plants_mart", dataset="athaliana_eg_gene", host="plants.ensembl.org")
listAttributes(mymart) # List available features
getBM(attributes=c("ensembl_gene_id", "description"), mart=mymart)[1:4,]
Efficient Sequence Parsing
getSeq
The following parses all annotation ranges provided by a GRanges object (e.g. gff) from a genome sequence stored in a local file.
gff <- gff[values(gff)$type != "chromosome"] # Remove chromosome ranges
rand <- DNAStringSet(sapply(unique(as.character(seqnames(gff))), function(x) paste(sample(c("A","T","G","C"), 200000, replace=T), collapse="")))
writeXStringSet(DNAStringSet(rand), "./data/test")
getSeq(FaFile("./data/test"), gff)
## DNAStringSet object of length 442:
## width seq names
## [1] 2269 GCCAGGTAGAATTCCAAAAATTGAAAGGCGTT...TGATCTGCCAGCGCCATTCGCTACGTCCGAAC Chr1
## [2] 2269 GCCAGGTAGAATTCCAAAAATTGAAAGGCGTT...TGATCTGCCAGCGCCATTCGCTACGTCCGAAC Chr1
## [3] 1871 CCTCCAAGTCTTAAAGCCTGCCTGCGACATTC...AAACCGCGAAATCTTATCGACCATTGTGTCTC Chr1
## [4] 283 GCCAGGTAGAATTCCAAAAATTGAAAGGCGTT...GCGGTCACTTTGCTAGGGCTGGTAGCCATGCA Chr1
## [5] 129 GCCAGGTAGAATTCCAAAAATTGAAAGGCGTT...TATCACACCTCTCTCTACTCGAATGACACGGC Chr1
## ... ... ...
## [438] 324 CTGGGTTGGAATGGGGCAAATACTTTCAAGGA...CAGGTGCAAATTGGCATCGCCGCACGGACGTT ChrM
## [439] 324 CTGGGTTGGAATGGGGCAAATACTTTCAAGGA...CAGGTGCAAATTGGCATCGCCGCACGGACGTT ChrM
## [440] 324 CTGGGTTGGAATGGGGCAAATACTTTCAAGGA...CAGGTGCAAATTGGCATCGCCGCACGGACGTT ChrM
## [441] 324 CTGGGTTGGAATGGGGCAAATACTTTCAAGGA...CAGGTGCAAATTGGCATCGCCGCACGGACGTT ChrM
## [442] 324 CTGGGTTGGAATGGGGCAAATACTTTCAAGGA...CAGGTGCAAATTGGCATCGCCGCACGGACGTT ChrM
extractTranscriptSeqs
Sequences composed of several ranges, such as transcripts (or CDSs) with several exons, can be parsed with extractTranscriptSeqs.
Note: the following expects the genome sequence in a file with path data/test and a valid txdb defining the ranges for that
genome.
library(GenomicFeatures); library(Biostrings); library(Rsamtools)
txdb <- loadDb("./data/TAIR10.sqlite")
indexFa("data/test") # Creates index for genome fasta
## [1] "data/test.fai"
txseq <- extractTranscriptSeqs(FaFile("data/test"), txdb, use.names=TRUE)
txseq
## DNAStringSet object of length 28:
## width seq names
## [1] 1688 GCCAGGTAGAATTCCAAAAATTGAAAGGCGTTT...TGATCTGCCAGCGCCATTCGCTACGTCCGAAC AT1G01010.1
## [2] 1623 CGCTTGTCCGTTCAAGAAAAGAATCTATTCCCC...CCACGCGTAGATTAGTTGGGGACGTAAGGACC AT1G01020.1
## [3] 1085 CGCTTGTCCGTTCAAGAAAAGAATCTATTCCCC...GAGCAGGTTCGTTTTTAGAGACTCTATACCGT AT1G01020.2
## [4] 1905 GTCCAGCCTACCCTATCGATACCTGTCACCATT...CAAAATCCGGCAGCATTTATCATGATCACGTA AT1G01030.1
## [5] 1239 CGCGGGAGAAACATTCGTCGAGGAATGATGCTG...ATGGTTTGCTCGACGAAGCTTATTCTGTCCGC AT2G01008.1
## ... ... ...
## [24] 1062 GCCGTCCACAATGACGGTGTGGATCTGAGAACG...TGGGTATCAGCCTACCGGCTGTAGGGAAATAG ATCG00020.1
## [25] 72 CCCGAGACTAGCCGCACCACGTGAGCTTTGGTG...AAACATCCTGTGCACGTTTTTTTGCCGGCGAT ATCG00030.1
## [26] 324 CTGGGTTGGAATGGGGCAAATACTTTCAAGGAG...CAGGTGCAAATTGGCATCGCCGCACGGACGTT ATMG00030.1
## [27] 462 GGATCTGACGCCACCAGAACCACAACTCCTGGA...GTGCTATGATGGCGTATGAGCGGGGGGTCGTA ATMG00010.1
## [28] 2568 CCGCTGATAGAGGTCAAATTACCAACAATAGTG...TGATAAATGGGATGCTTTAAGGACTCGTCGGC ATMG00020.1
Homework 6
See here.
Session Info
sessionInfo()
## R version 4.1.3 (2022-03-10)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Debian GNU/Linux 10 (buster)
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.8.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.8.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
## [4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] grid stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] GenomicFeatures_1.46.1 AnnotationDbi_1.56.2 rtracklayer_1.54.0
## [4] systemPipeR_2.0.4 ShortRead_1.52.0 GenomicAlignments_1.30.0
## [7] SummarizedExperiment_1.24.0 Biobase_2.54.0 MatrixGenerics_1.6.0
## [10] matrixStats_0.61.0 Rsamtools_2.10.0 GenomicRanges_1.46.1
## [13] BiocParallel_1.28.2 ggseqlogo_0.1 ggplot2_3.3.5
## [16] seqLogo_1.60.0 Biostrings_2.62.0 GenomeInfoDb_1.30.0
## [19] XVector_0.34.0 IRanges_2.28.0 S4Vectors_0.32.3
## [22] BiocGenerics_0.40.0 BiocStyle_2.22.0
##
## loaded via a namespace (and not attached):
## [1] bitops_1.0-7 bit64_4.0.5 filelock_1.0.2 progress_1.2.2
## [5] RColorBrewer_1.1-2 httr_1.4.2 tools_4.1.3 bslib_0.3.1
## [9] utf8_1.2.2 R6_2.5.1 DBI_1.1.1 colorspace_2.0-2
## [13] withr_2.4.3 prettyunits_1.1.1 tidyselect_1.1.1 curl_4.3.2
## [17] bit_4.0.4 compiler_4.1.3 cli_3.1.0 xml2_1.3.3
## [21] DelayedArray_0.20.0 labeling_0.4.2 bookdown_0.24 sass_0.4.0
## [25] scales_1.1.1 rappdirs_0.3.3 stringr_1.4.0 digest_0.6.29
## [29] rmarkdown_2.13 jpeg_0.1-9 pkgconfig_2.0.3 htmltools_0.5.2
## [33] highr_0.9 dbplyr_2.1.1 fastmap_1.1.0 htmlwidgets_1.5.4
## [37] rlang_1.0.2 RSQLite_2.2.9 farver_2.1.0 jquerylib_0.1.4
## [41] BiocIO_1.4.0 generics_0.1.1 hwriter_1.3.2 jsonlite_1.8.0
## [45] dplyr_1.0.7 RCurl_1.98-1.5 magrittr_2.0.2 GenomeInfoDbData_1.2.7
## [49] Matrix_1.4-0 Rcpp_1.0.8.2 munsell_0.5.0 fansi_0.5.0
## [53] lifecycle_1.0.1 stringi_1.7.6 yaml_2.3.5 zlibbioc_1.40.0
## [57] BiocFileCache_2.2.0 blob_1.2.2 parallel_4.1.3 crayon_1.4.2
## [61] lattice_0.20-45 hms_1.1.1 KEGGREST_1.34.0 knitr_1.37
## [65] pillar_1.6.4 rjson_0.2.20 codetools_0.2-18 biomaRt_2.50.1
## [69] XML_3.99-0.8 glue_1.6.2 evaluate_0.15 blogdown_1.8.2
## [73] latticeExtra_0.6-29 BiocManager_1.30.16 png_0.1-7 vctrs_0.3.8
## [77] gtable_0.3.0 purrr_0.3.4 assertthat_0.2.1 cachem_1.0.6
## [81] xfun_0.30 restfulr_0.0.13 viridisLite_0.4.0 tibble_3.1.6
## [85] memoise_2.0.1 ellipsis_0.3.2
References
Huber, Wolfgang, Vincent J Carey, Robert Gentleman, Simon Anders, Marc Carlson, Benilton S Carvalho, Hector Corrada Bravo, et al. 2015. “Orchestrating High-Throughput Genomic Analysis with Bioconductor.” Nat. Methods 12 (2): 115–21. https://doi.org/10.1038/nmeth.3252.
Lawrence, Michael, Wolfgang Huber, Hervé Pagès, Patrick Aboyoun, Marc Carlson, Robert Gentleman, Martin T Morgan, and Vincent J Carey. 2013. “Software for Computing and Annotating Genomic Ranges.” PLoS Comput. Biol. 9 (8): e1003118. https://doi.org/10.1371/journal.pcbi.1003118.